
Uncovering the Contribution of Moderate-Penetrance Susceptibility Genes to Breast Cancer by Whole-Exome Sequencing and Targeted Enrichment Sequencing of Candidate Genes in Women of European Ancestry

 , , , , , , , , , ,
, , , , , , , , , ,  , , , , , , , , , , ,
, , , , , , , , , , ,  , add
Show full author list
, add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Discovery Stage—Whole-Exome Sequencing (WES) Analysis
2.1.1. Studies and Datasets
2.1.2. Selection of Breast Cancer Cases
2.1.3. Library Preparation, High-Throughput Sequencing, and Bioinformatics Analysis
2.1.4. Variant Filtering and Gene Prioritization
2.1.5. Aggregation of Gene Lists
2.2. Validation Stage—Targeted Enrichment Sequencing
2.2.1. Sample Sets
2.2.2. Power Calculations
2.2.3. Library Preparation and High-Throughput Sequencing
2.2.4. Bioinformatics and Statistical Analyses
3. Results
3.1. Known or Suspected Breast Cancer Susceptibility Genes
3.1.1. Loss-of-Function Variants
3.1.2. Rare Missense Variants
3.2. Validation of Candidate Genes Identified at the Discovery Stage
3.2.1. Loss-of-Function Variants
3.2.2. Rare Missense Variants
3.2.3. Combined Analysis of LoF and Rare Missense Variants
3.2.4. Individual LoF and Missense Variant Analysis
4. Discussion
Limitations and Weaknesses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiezun, A.; Garimella, K.; Do, R.; Stitziel, N.O.; Neale, B.M.; McLaren, P.J.; Gupta, N.; Sklar, P.; Sullivan, P.F.; Moran, J.L.; et al. Exome sequencing and the genetic basis of complex traits. Nat. Genet. 2012, 44, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Chandler, M.R.; Bilgili, E.P.; Merner, N.D. A Review of whole-exome sequencing efforts toward hereditary breast cancer susceptibility gene discovery. Hum. Mutat. 2016, 37, 835–846. [Google Scholar] [CrossRef]
- Zelli, V.; Compagnoni, C.; Cannita, K.; Capelli, R.; Capalbo, C.; Di Vito Nolfi, M.; Alesse, E.; Zazzeroni, F.; Tessitore, A. Applications of Next Generation Sequencing to the Analysis of Familial Breast/Ovarian Cancer. High Throughput 2020, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers with Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2019, 5, 51–57. [Google Scholar] [CrossRef]
- Kast, K.; Rhiem, K.; Wappenschmidt, B.; Hahnen, E.; Hauke, J.; Bluemcke, B.; Zarghooni, V.; Herold, N.; Ditsch, N.; Kiechle, M.; et al. Prevalence of BRCA1/2 germline mutations in 21,401 families with breast and ovarian cancer. J. Med. Genet. 2016, 53, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.K.; Hogervorst, F.; van Hien, R.; Cornelissen, S.; Broeks, A.; Adank, M.A.; Meijers, H.; Waisfisz, Q.; Hollestelle, A.; Schutte, M.; et al. Age- and Tumor Subtype-Specific Breast Cancer Risk Estimates for CHEK2*1100delC Carriers. J. Clin. Oncol. 2016, 34, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Kriege, M.; Hollestelle, A.; Jager, A.; Huijts, P.E.; Berns, E.M.; Sieuwerts, A.M.; Meijer-van Gelder, M.E.; Collée, J.M.; Devilee, P.; Hooning, M.J.; et al. Survival and contralateral breast cancer in CHEK2 1100delC breast cancer patients: Impact of adjuvant chemotherapy. Br. J. Cancer 2014, 111, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.K.; Tollenaar, R.A.; de Kemp, S.R.; Broeks, A.; Cornelisse, C.J.; Smit, V.T.; Peterse, J.L.; van Leeuwen, F.E.; Van’t Veer, L.J. Breast cancer survival and tumor characteristics in premenopausal women carrying the CHEK2*1100delC germline mutation. J. Clin. Oncol. 2007, 25, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.A.; Brusselle, G.; Ghanbari, M.; Goedegebure, A.; Ikram, M.K.; Kavousi, M.; Kieboom, B.C.T.; Klaver, C.C.W.; de Knegt, R.J.; Luik, A.I.; et al. Objectives, design and main findings until 2020 from the Rotterdam Study. Eur. J. Epidemiol. 2020, 35, 483–517. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Gurvey, V.; Vermaat, M.; Van Dijrk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2013, 22, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014, 46, 818–825. [Google Scholar] [CrossRef]
- Jessen, F.; Wiese, B.; Bickel, H.; Eiffländer-Gorfer, S.; Fuchs, A.; Kaduszkiewicz, H.; Köhler, M.; Luck, T.; Mösch, E.; Pentzek, M.; et al. Prediction of dementia in primary care patients. PLoS ONE 2011, 6, e16852. [Google Scholar] [CrossRef]
- Wichmann, H.E.; Gieger, C.; Illig, T.; MONICA/KORA Study Group. KORA-gen-resource for population genetics, controls and a broad spectrum of disease phenotypes. Das Gesundh. 2005, 67, S26–S30. [Google Scholar] [CrossRef] [Green Version]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Liu, J.; Prager-van der Smissen, W.J.C.; Schmidt, M.K.; Collée, J.M.; Cornelissen, S.; Lamping, R.; Nieuwlaat, A.; Foekens, J.A.; Hooning, M.J.; Verhoef, S.; et al. Recurrent HOXB13 mutations in the Dutch population do not associate with increased breast cancer risk. Sci. Rep. 2016, 6, 30026. [Google Scholar] [CrossRef] [Green Version]
- Dunning, A.M.; Healey, C.S.; Pharoah, P.D.; Teare, M.D.; Ponder, B.A.; Easton, D.F. A systematic review of genetic polymorphisms and breast Cancer risk. Cancer Epidemiol. Biomark. Prev. 1999, 8, 843–854. [Google Scholar]
- Day, N.; Oakes, S.; Luben, R.; Khaw, K.T.; Bingham, S.; Welch, A.; Wareham, N. EPIC-Norfolk: Study design and characteristics of the cohort. european prospective investigation of Cancer. Br. J. Cancer 1999, 80, 95–103. [Google Scholar] [PubMed]
- Kataoka, M.; Antoniou, A.; Warren, R.; Leyland, J.; Brown, J.; Audley, T.; Easton, D. Genetic models for the familial aggregation of mammographic breast density. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1277–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, E.M.; Hopper, J.L.; Beck, J.C.; Knight, J.A.; Neuhausen, S.L.; Senie, R.T.; Ziogas, A.; Andrulis, I.L.; Anton-Culver, H.; Boyd, N.; et al. The Breast Cancer Family Registry: An infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004, 6, R375–R389. [Google Scholar] [CrossRef] [Green Version]
- Awadalla, P.; Boileau, C.; Payette, Y.; Idaghdour, Y.; Goulet, J.P.; Knoppers, B.; Hamet, P.; Laberge, C.; CARTaGENE Project. Cohort profile of the CARTaGENE study: Quebec’s population-based biobank for public health and personalized genomics. Int. J. Epidemiol. 2013, 42, 1285–1299. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From Fastq Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Feng, B.J. PERCH: A unified framework for disease gene prioritization. Hum. Mutat. 2017, 38, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Abecasis, G.R.; Boehnke, M.; Lin, X. Rare-variant association analysis: Study designs and statistical tests. Am. J. Hum. Genet. 2014, 95, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.J.; Peloso, G.M.; Zhan, X.; Holmen, O.L.; Zawistowski, M.; Shuang, F.; Nikpay, M.; Auer, P.L.; Goel, A.; Zhang, H.; et al. Meta-analysis of gene-level tests for rare variant association. Nat. Genet. 2014, 46, 200–204. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic. Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhu, D.; Zhang, X.; Zhang, Y.; Zhou, J.; Dong, M. TMEM206 promotes the malignancy of colorectal cancer cells by interacting with AKT and extracellular signal-regulated kinase signaling pathways. J. Cell. Physiol. 2019, 234, 10888–10898. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.S.; Fujimoto, J.; Tamaya, T. Clinical implications of coexpression of growth arrest-specific gene 6 and receptor tyrosine kinases Axl and Sky in human uterine leiomyoma. Mol. Hum. Reprod. 2003, 9, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Wurdak, H.; Wang, Y.; Galkin, A.; Tao, H.; Li, J.; Lyssiotis, C.A.; Yan, F.; Tu, B.P.; Miraglia, L.; et al. A genomic screen identifies TYRO3 as a MITF regulator in melanoma. Proc. Natl. Acad. Sci. USA 2009, 106, 17025–17030. [Google Scholar] [CrossRef] [Green Version]
- Shahi, R.B.; De Brakeleer, S.; Caljon, B.; Pauwels, I.; Bonduelle, M.; Joris, S.; Fontaine, C.; Vanhoeij, M.; Van Dooren, S.; Teugels, E.; et al. Identification of candidate cancer predisposing variants by performing whole-exome sequencing on index patients from BRCA1 and BRCA2-negative breast cancer families. BMC Cancer 2019, 19, 313. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Bakshi, D.; Sharma, V.; Sharma, I.; Shah, R.; Bhat, A.; Bhat, G.R.; Bhanu Sharma, B.; Wakhloo, A.; Kaul, S.; et al. Genetic variants of DNAH11 and LRFN2 genes and their association with ovarian and breast cancer. Int. J. Gynaecol. Obs. 2020, 148, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Navarro, P.; González-Neira, A.; Pita, G.; Díaz-Uriarte, R.; Tais Moreno, L.; Ederra, M.; Pedraz-Pingarrón, C.; Sánchez-Contador, C.; Antonio Vázquez-Carrete, J.; Moreo, P.; et al. Genome wide association study identifies a novel putative mammographic density locus at 1q12-q21. Int. J. Cancer 2015, 136, 2427–2436. [Google Scholar] [CrossRef] [PubMed]
- Fachal, L.; Aschard, H.; Beesley, J.; Barnes, D.R.; Allen, J.; Kar, S.; Pooley, K.A.; Dennis, J.; Michailidou, K.; Turman, C.; et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat. Genet. 2020, 52, 56–73. [Google Scholar] [CrossRef]
- Lucci, M.A.; Orlandi, R.; Triulzi, T.; Tagliabue, E.; Balsari, A.; Villa-Moruzzi, E. Expression profile of tyrosine phosphatases in HER2 breast cancer cells and tumors. Cell. Oncol. 2010, 32, 361–372. [Google Scholar] [CrossRef]
- Wu, K.-G.; He, X.-F.; Li, Y.-H.; Xie, W.-B.; Huang, X. Association between the XPD/ERCC2 Lys751Gln polymorphism and risk of cancer: Evidence from 224 case-control studies. Tumour Biol. 2014, 35, 11243–11259. [Google Scholar] [CrossRef]
- Rump, A.; Benet-Pages, A.; Schubert, S.; Kuhlmann, J.D.; Janavičius, R.; Macháčková, E.; Foretová, L.; Kleibl, Z.; Lhota, F.; Zemankova, P.; et al. Identification and Functional Testing of ERCC2 Mutations in a Multi-national Cohort of Patients with Familial Breast- and Ovarian Cancer. PLoS Genet. 2016, 12, e1006248. [Google Scholar] [CrossRef] [PubMed]
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.R.; Rowley, S.M.; Li, N.; McInerny, S.; Devereux, L.; Wong-Brown, M.W.; Trainer, A.H.; Mitchell, G.; Scott, R.J.; James, P.A.; et al. Panel Testing for Familial Breast Cancer: Calibrating the Tension between Research and Clinical Care. J. Clin. Oncol. 2016, 34, 1455–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloraifi, F.; McCartan, D.; McDevitt, T.; Green, A.J.; Bracken, A.; Geraghty, J. Protein-truncating variants in moderate-risk breast cancer susceptibility genes: A meta-analysis of high-risk case-control screening studies. Cancer Genet. 2015, 208, 455–463. [Google Scholar] [CrossRef]
- Lee, K.; Seifert, B.A.; Shimelis, H.; Ghosh, R.; Crowley, S.B.; Carter, N.J.; Doonanco, K.; Foreman, A.K.; Ritter, D.I.; Jimenez, S.; et al. Clinical validity assessment of genes frequently tested on hereditary breast and ovarian cancer susceptibility sequencing panels. Genet. Med. 2019, 21, 1497–1506. [Google Scholar] [CrossRef]
- Ku, C.S.; Cooper, D.N.; Patrinos, G.P. The Rise and Rise of Exome Sequencing. Public Health Genom. 2016, 19, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Lelieveld, S.H.; Spielmann, M.; Mundlos, S.; Veltman, J.A.; Gilissen, C. Comparison of Exome and Genome Sequencing Technologies for the Complete Capture of Protein-Coding Regions. Hum. Mutat. 2015, 36, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Wu, L.; Schaid, D.J.; Sicotte, H.; Wieben, E.D.; Li, H.; Petersen, G.M. Case-only exome sequencing and complex disease susceptibility gene discovery: Study design considerations. J. Med. Genet. 2015, 52, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Gusev, A.; Lee, S.H.; Trynka, G.; Finucane, H.; Vilhjálmsson, B.J.; Xu, H.; Zang, C.; Ripke, S.; Bulik-Sullivan, B.; Stahl, E.; et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 2014, 95, 535–552. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics 2013, 1303, 3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, A.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP V3.0: A One-Stop Database of Functional Predictions and Annotations for Hu-man Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the World’s Knowledge of Somatic Mutations in Human Cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the Sift Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using Polyphen-2. Curr. Protoc. Hum. Genet. 2013, 7, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimand, J.; Bader, G.D. Systematic Analysis of Somatic Mutations in Phosphorylation Signaling Predicts Novel Cancer Driv-ers. Mol. Syst. Biol. 2013, 9, 637. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Overall | ER-Negative | ER-Positive | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Number of Carriers | Number of Carriers | OR | 95% CI | p-Value | Number of Carriers | OR | 95% CI | p-Value | Number of Carriers | OR | 95% CI | p-Value | |||
| Controls (n = 6019) | Cases (n = 6211) | Cases (n = 808) | Cases (n = 2764) | |||||||||||||
| AKT1 | 1 | 1 | 0.89 | 0.06 | 14.25 | 0.935 | 0 | 0.00 | 0.00 | Inf | 0.997 | 1 | 1.74 | 0.11 | 27.90 | 0.694 |
| ATM | 20 | 74 | 3.55 | 2.16 | 5.82 | 5.44 × 10−7 | 3 | 0.89 | 0.26 | 3.02 | 0.851 | 42 | 4.06 | 2.37 | 6.97 | 3.39 × 10−7 |
| BARD1 | 4 | 11 | 2.54 | 0.81 | 7.98 | 0.110 | 2 | 2.98 | 0.54 | 16.57 | 0.212 | 7 | 3.13 | 0.91 | 10.71 | 0.069 |
| BRE/BABAM2 | 0 | 0 | - | - | - | - | 0 | - | - | - | - | 0 | - | - | - | - |
| BRIP1 | 8 | 14 | 1.64 | 0.69 | 3.93 | 0.262 | 2 | 1.44 | 0.30 | 6.84 | 0.649 | 7 | 1.78 | 0.64 | 4.97 | 0.274 |
| CDH1 | 0 | 4 | 0.00 | 0.00 | Inf | 0.906 | 2 | 0.00 | 0.00 | Inf | 0.994 | 1 | 0.00 | 0.00 | Inf | 0.994 |
| CHEK2 | 32 | 111 | 3.47 | 2.33 | 5.15 | 7.62 × 10−10 | 9 | 2.65 | 1.24 | 5.70 | 0.012 | 56 | 4.44 | 2.82 | 6.99 | 1.23 × 10−10 |
| EPCAM | 3 | 1 | 0.33 | 0.04 | 3.22 | 0.343 | 0 | 0.00 | Inf | 0.995 | 1 | 0.60 | 0.06 | 5.77 | 0.658 | |
| FAM175A/ABRAXAS1 | 0 | 0 | - | - | - | - | 0 | - | - | - | - | 0 | - | - | - | - |
| FANCC | 15 | 13 | 0.86 | 0.41 | 1.81 | 0.692 | 1 | 0.63 | 0.08 | 4.91 | 0.662 | 6 | 1.00 | 0.37 | 2.66 | 0.995 |
| FANCM | 39 | 42 | 1.03 | 0.67 | 1.60 | 0.894 | 8 | 1.52 | 0.70 | 3.30 | 0.295 | 20 | 1.09 | 0.63 | 1.90 | 0.749 |
| GEN1 | 2 | 3 | 1.54 | 0.26 | 9.26 | 0.636 | 1 | 7.68 | 0.45 | 131.19 | 0.159 | 2 | 3.56 | 0.32 | 39.34 | 0.300 |
| MEN1 | 0 | 0 | - | - | - | - | 0 | - | - | - | - | 0 | - | - | - | - |
| MLH1 | 1 | 1 | 0.89 | 0.06 | 14.25 | 0.935 | 0 | 0.00 | Inf | 0.997 | 1 | 1.74 | 0.11 | 27.90 | 0.694 | |
| MRE11A/MRE11 | 0 | 0 | - | - | - | - | 0 | - | - | - | - | 0 | - | - | - | - |
| MSH2 | 0 | 2 | 0.00 | 0.00 | Inf | 0.934 | 0 | - | - | - | - | 1 | 0.00 | 0.00 | Inf | 0.994 |
| MSH6 | 47 | 19 | 0.36 | 0.21 | 0.62 | 2.07 × 10−4 | 5 | 0.55 | 0.22 | 1.39 | 0.204 | 11 | 0.42 | 0.22 | 0.81 | 9.00 × 10−3 |
| MUTYH | 5 | 4 | 0.78 | 0.21 | 2.93 | 0.718 | 1 | 2.06 | 0.21 | 20.54 | 0.537 | 1 | 0.73 | 0.07 | 7.19 | 0.784 |
| NBN | 27 | 19 | 0.65 | 0.36 | 1.17 | 0.150 | 3 | 0.61 | 0.18 | 2.02 | 0.416 | 13 | 0.90 | 0.46 | 1.76 | 0.766 |
| NF1 | 18 | 19 | 0.95 | 0.50 | 1.81 | 0.867 | 3 | 0.86 | 0.25 | 2.92 | 0.805 | 13 | 1.26 | 0.62 | 2.59 | 0.522 |
| PALB2 | 10 | 42 | 3.95 | 1.98 | 7.88 | 9.68 × 10−5 | 7 | 6.45 | 2.31 | 17.98 | 3.72 × 10−4 | 22 | 5.27 | 2.39 | 11.61 | 3.83 × 10−5 |
| PIK3CA | 0 | 0 | - | - | - | - | 0 | - | - | - | - | 0 | - | - | - | - |
| PMS2 | 27 | 16 | 0.53 | 0.29 | 0.98 | 0.044 | 3 | 0.57 | 0.17 | 1.87 | 0.351 | 8 | 0.53 | 0.24 | 1.16 | 0.111 |
| PTEN | 1 | 2 | 1.78 | 0.16 | 19.65 | 0.637 | 0 | 0.00 | 0.00 | Inf | 0.997 | 1 | 1.74 | 0.11 | 27.90 | 0.694 |
| RAD50 | 9 | 9 | 0.97 | 0.38 | 2.44 | 0.943 | 1 | 1.05 | 0.13 | 8.59 | 0.967 | 8 | 2.45 | 0.90 | 6.71 | 0.081 |
| RAD51C | 3 | 6 | 1.89 | 0.47 | 7.55 | 0.370 | 2 | 3.90 | 0.64 | 23.88 | 0.141 | 2 | 1.19 | 0.20 | 7.14 | 0.847 |
| RAD51D | 3 | 7 | 2.18 | 0.56 | 8.45 | 0.258 | 5 | 11.26 | 2.60 | 48.68 | 1.19 × 10−3 | 1 | 0.74 | 0.08 | 7.31 | 0.795 |
| RECQL | 9 | 12 | 1.28 | 0.54 | 3.05 | 0.576 | 0 | 0.00 | 0.00 | Inf | 0.990 | 6 | 1.64 | 0.55 | 4.92 | 0.379 |
| RINT1 | 6 | 5 | 0.76 | 0.23 | 2.50 | 0.655 | 0 | 0.00 | 0.00 | Inf | 0.992 | 2 | 0.59 | 0.12 | 2.90 | 0.512 |
| STK11 | 0 | 0 | - | - | - | - | 0 | - | - | - | - | 0 | - | - | - | - |
| TP53 | 1 | 11 | 10.16 | 1.31 | 78.67 | 0.026 | 1 | 5.00 | 0.31 | 80.03 | 0.255 | 3 | 5.24 | 0.54 | 50.39 | 0.152 |
| XRCC2 | 0 | 0 | - | - | - | - | 0 | - | - | - | - | 0 | - | - | - | - |
| Overall | ER-Negative | ER-Positive | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Carriers | OR | 95% CI | p-Value | Number of Carriers | OR | 95% CI | p-Value | Number of Carriers | OR | 95% CI | p-Value | |||||
| Controls n = 6019 | Cases n = 6211 | Cases n = 808 | Cases n = 2764 | |||||||||||||
| (A) Loss-of-function variants | ||||||||||||||||
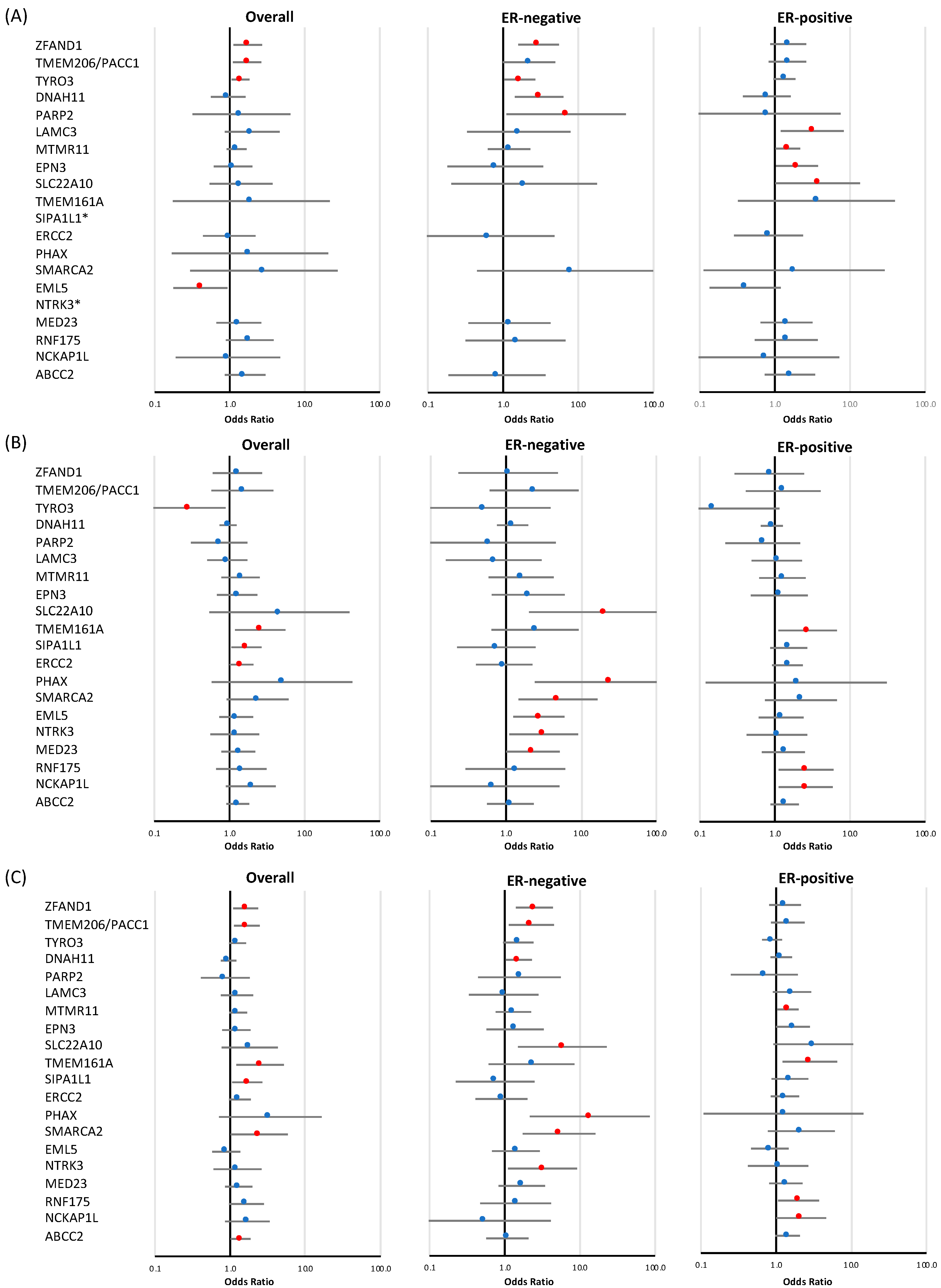
| ZFAND1 | 31 | 59 | 1.73 | 1.12 | 2.68 | 0.014 | 16 | 2.96 | 1.59 | 5.50 | 6.37 × 10−4 | 24 | 1.50 | 0.87 | 2.60 | 0.146 |
| TMEM206/PACC1 | 33 | 56 | 1.70 | 1.11 | 2.62 | 0.016 | 8 | 2.21 | 0.99 | 4.93 | 0.052 | 21 | 1.47 | 0.83 | 2.61 | 0.186 |
| TYRO3 | 89 | 131 | 1.40 | 1.06 | 1.83 | 0.016 | 22 | 1.66 | 1.03 | 2.68 | 0.038 | 58 | 1.34 | 0.95 | 1.88 | 0.092 |
| DNAH11 | 28 | 27 | 0.96 | 0.56 | 1.62 | 0.865 | 10 | 3.00 | 1.42 | 6.32 | 3.88 × 10−3 | 10 | 0.78 | 0.37 | 1.63 | 0.507 |
| PARP2 | 3 | 4 | 1.43 | 0.32 | 6.40 | 0.641 | 2 | 6.89 | 1.11 | 42.78 | 0.038 | 1 | 0.75 | 0.08 | 7.45 | 0.808 |
| LAMC3 | 8 | 17 | 2.00 | 0.86 | 4.64 | 0.107 | 2 | 1.61 | 0.33 | 7.87 | 0.553 | 11 | 3.14 | 1.20 | 8.20 | 0.020 |
| MTMR11 | 75 | 95 | 1.24 | 0.91 | 1.68 | 0.174 | 11 | 1.20 | 0.63 | 2.29 | 0.583 | 49 | 1.49 | 1.03 | 2.17 | 0.037 |
| EPN3 | 21 | 24 | 1.10 | 0.61 | 1.99 | 0.742 | 2 | 0.79 | 0.18 | 3.40 | 0.747 | 18 | 1.97 | 1.03 | 3.76 | 0.039 |
| SLC22A10 | 7 | 10 | 1.41 | 0.54 | 3.72 | 0.484 | 1 | 1.91 | 0.21 | 17.73 | 0.571 | 6 | 3.70 | 1.01 | 13.57 | 0.048 |
| TMEM161A | 1 | 2 | 1.94 | 0.18 | 21.43 | 0.590 | 0 | 0.00 | 0.00 | Inf | 0.997 | 2 | 3.56 | 0.32 | 39.34 | 0.300 |
| SIPA1L1 | 0 | 0 | - | - | - | - | 0 | - | - | - | - | 0 | - | - | - | - |
| ERCC2 | 12 | 12 | 0.99 | 0.44 | 2.20 | 0.972 | 1 | 0.62 | 0.08 | 4.83 | 0.646 | 5 | 0.83 | 0.29 | 2.38 | 0.724 |
| PHAX | 1 | 2 | 1.86 | 0.17 | 20.52 | 0.612 | 0 | 0.00 | 0.00 | Inf | 0.997 | 0 | 0.00 | Inf | 0.995 | |
| SMARCA2 | 1 | 3 | 2.85 | 0.30 | 27.42 | 0.365 | 1 | 7.68 | 0.45 | 131.19 | 0.159 | 1 | 1.80 | 0.11 | 28.81 | 0.678 |
| EML5 | 18 | 8 | 0.41 | 0.18 | 0.94 | 0.035 | 0 | 0.00 | 0.00 | Inf | 0.986 | 4 | 0.40 | 0.14 | 1.19 | 0.100 |
| NTRK3 | 0 | 1 | 0.00 | 0.00 | inf | 0.930 | 0 | - | - | - | - | 0 | - | - | - | - |
| MED23 | 14 | 20 | 1.32 | 0.67 | 2.62 | 0.424 | 3 | 1.21 | 0.35 | 4.27 | 0.763 | 11 | 1.43 | 0.65 | 3.17 | 0.375 |
| RNF175 | 11 | 21 | 1.85 | 0.89 | 3.85 | 0.099 | 2 | 1.46 | 0.32 | 6.75 | 0.628 | 7 | 1.42 | 0.54 | 3.72 | 0.478 |
| NCKAP1L | 3 | 3 | 0.95 | 0.19 | 4.71 | 0.948 | 0 | 0.00 | 0.00 | Inf | 0.995 | 1 | 0.73 | 0.07 | 7.19 | 0.784 |
| ABCC2 | 16 | 26 | 1.60 | 0.86 | 2.99 | 0.140 | 2 | 0.83 | 0.19 | 3.67 | 0.804 | 12 | 1.59 | 0.74 | 3.42 | 0.240 |
| (B) Rare missense variants | ||||||||||||||||
| ZFAND1 | 12 | 16 | 1.27 | 0.60 | 2.70 | 0.527 | 2 | 1.07 | 0.23 | 4.89 | 0.930 | 5 | 0.85 | 0.29 | 2.45 | 0.759 |
| TMEM206/PACC1 | 7 | 11 | 1.48 | 0.57 | 3.83 | 0.416 | 3 | 2.37 | 0.61 | 9.27 | 0.214 | 5 | 1.29 | 0.41 | 4.06 | 0.669 |
| TYRO3 | 13 | 4 | 0.29 | 0.10 | 0.89 | 0.031 | 1 | 0.51 | 0.07 | 3.93 | 0.516 | 1 | 0.15 | 0.02 | 1.15 | 0.068 |
| DNAH11 | 116 | 115 | 0.95 | 0.74 | 1.24 | 0.726 | 21 | 1.22 | 0.76 | 1.97 | 0.406 | 52 | 0.91 | 0.65 | 1.28 | 0.603 |
| PARP2 | 12 | 9 | 0.73 | 0.31 | 1.73 | 0.470 | 1 | 0.59 | 0.08 | 4.61 | 0.614 | 4 | 0.69 | 0.22 | 2.17 | 0.524 |
| LAMC3 | 21 | 20 | 0.93 | 0.51 | 1.73 | 0.826 | 2 | 0.69 | 0.16 | 3.00 | 0.620 | 10 | 1.06 | 0.49 | 2.30 | 0.888 |
| MTMR11 | 19 | 28 | 1.40 | 0.78 | 2.52 | 0.256 | 5 | 1.59 | 0.59 | 4.31 | 0.362 | 13 | 1.26 | 0.62 | 2.56 | 0.524 |
| EPN3 | 18 | 23 | 1.26 | 0.68 | 2.35 | 0.460 | 4 | 1.98 | 0.65 | 6.05 | 0.233 | 8 | 1.15 | 0.48 | 2.74 | 0.751 |
| SLC22A10 | 1 | 5 | 4.61 | 0.54 | 39.46 | 0.163 | 3 | 20.76 | 2.03 | 211.95 | 0.011 | 0 | 0.00 | 0.00 | Inf | 0.995 |
| TMEM161A | 9 | 24 | 2.56 | 1.19 | 5.51 | 0.016 | 3 | 2.44 | 0.64 | 9.26 | 0.190 | 11 | 2.73 | 1.11 | 6.71 | 0.029 |
| SIPA1L1 | 29 | 50 | 1.68 | 1.06 | 2.67 | 0.026 | 3 | 0.74 | 0.22 | 2.48 | 0.631 | 22 | 1.53 | 0.87 | 2.69 | 0.140 |
| ERCC2 | 49 | 73 | 1.45 | 1.01 | 2.08 | 0.047 | 6 | 0.95 | 0.40 | 2.25 | 0.906 | 31 | 1.47 | 0.92 | 2.35 | 0.108 |
| PHAX | 1 | 5 | 4.99 | 0.58 | 42.78 | 0.143 | 3 | 24.27 | 2.42 | 243.37 | 6.69 × 10−3 | 1 | 1.92 | 0.12 | 30.68 | 0.646 |
| SMARCA2 | 6 | 15 | 2.36 | 0.92 | 6.09 | 0.076 | 5 | 4.92 | 1.47 | 16.48 | 9.69 × 10−3 | 7 | 2.23 | 0.74 | 6.71 | 0.153 |
| EML5 | 26 | 33 | 1.23 | 0.73 | 2.06 | 0.432 | 9 | 2.74 | 1.25 | 6.01 | 0.012 | 13 | 1.21 | 0.61 | 2.41 | 0.587 |
| NTRK3 | 13 | 15 | 1.18 | 0.56 | 2.48 | 0.669 | 5 | 3.17 | 1.11 | 9.11 | 0.032 | 7 | 1.06 | 0.42 | 2.69 | 0.897 |
| MED23 | 25 | 34 | 1.31 | 0.78 | 2.20 | 0.305 | 8 | 2.29 | 1.01 | 5.18 | 0.048 | 15 | 1.30 | 0.67 | 2.51 | 0.434 |
| RNF175 | 11 | 16 | 1.43 | 0.66 | 3.09 | 0.361 | 2 | 1.34 | 0.29 | 6.14 | 0.710 | 12 | 2.59 | 1.12 | 6.00 | 0.026 |
| NCKAP1L | 10 | 20 | 1.92 | 0.90 | 4.10 | 0.093 | 1 | 0.65 | 0.08 | 5.16 | 0.686 | 13 | 2.57 | 1.12 | 5.91 | 0.027 |
| ABCC2 | 57 | 76 | 1.29 | 0.91 | 1.82 | 0.150 | 9 | 1.14 | 0.56 | 2.34 | 0.714 | 36 | 1.36 | 0.88 | 2.09 | 0.163 |
| (C) Combined analysis: loss-of-function and rare missense variants | ||||||||||||||||
| ZFAND1 | 43 | 75 | 1.61 | 1.1 | 2.34 | 0.014 | 18 | 2.47 | 1.40 | 4.36 | 1.78 × 10−3 | 29 | 1.32 | 0.82 | 2.15 | 0.257 |
| TMEM206/PACC1 | 40 | 67 | 1.67 | 1.12 | 2.47 | 0.011 | 11 | 2.26 | 1.13 | 4.51 | 0.021 | 26 | 1.43 | 0.86 | 2.39 | 0.169 |
| TYRO3 | 102 | 135 | 1.25 | 0.97 | 1.63 | 0.090 | 23 | 1.51 | 0.95 | 2.41 | 0.082 | 59 | 1.18 | 0.85 | 1.64 | 0.324 |
| DNAH11 | 144 | 142 | 0.95 | 0.75 | 1.21 | 0.696 | 31 | 1.53 | 1.03 | 2.29 | 0.037 | 62 | 0.89 | 0.65 | 1.21 | 0.449 |
| PARP2 | 15 | 13 | 0.86 | 0.41 | 1.81 | 0.694 | 3 | 1.57 | 0.44 | 5.58 | 0.484 | 5 | 0.70 | 0.25 | 1.96 | 0.497 |
| LAMC3 | 29 | 37 | 1.23 | 0.76 | 2.01 | 0.402 | 4 | 0.96 | 0.33 | 2.80 | 0.947 | 21 | 1.63 | 0.91 | 2.93 | 0.104 |
| MTMR11 | 94 | 123 | 1.27 | 0.97 | 1.67 | 0.082 | 16 | 1.30 | 0.75 | 2.24 | 0.345 | 62 | 1.44 | 1.03 | 2.01 | 0.032 |
| EPN3 | 38 | 47 | 1.21 | 0.79 | 1.86 | 0.385 | 6 | 1.37 | 0.57 | 3.30 | 0.484 | 26 | 1.67 | 1.00 | 2.81 | 0.051 |
| SLC22A10 | 8 | 15 | 1.82 | 0.77 | 4.31 | 0.171 | 4 | 5.84 | 1.51 | 22.69 | 0.011 | 6 | 3.12 | 0.92 | 10.61 | 0.068 |
| TMEM161A | 10 | 26 | 2.50 | 1.2 | 5.19 | 0.014 | 3 | 2.26 | 0.61 | 8.44 | 0.224 | 13 | 2.83 | 1.22 | 6.55 | 0.015 |
| SIPA1L1 | 29 | 50 | 1.68 | 1.06 | 2.67 | 0.026 | 3 | 0.74 | 0.22 | 2.48 | 0.631 | 22 | 1.53 | 0.87 | 2.69 | 0.140 |
| ERCC2 | 60 | 83 | 1.34 | 0.96 | 1.88 | 0.083 | 7 | 0.90 | 0.41 | 2.01 | 0.802 | 35 | 1.32 | 0.86 | 2.04 | 0.210 |
| PHAX | 2 | 7 | 3.41 | 0.71 | 16.4 | 0.126 | 3 | 13.49 | 2.15 | 84.71 | 5.50 × 10−3 | 1 | 1.26 | 0.11 | 14.53 | 0.854 |
| SMARCA2 | 7 | 18 | 2.43 | 1.01 | 5.83 | 0.046 | 6 | 5.27 | 1.73 | 16.05 | 3.40 × 10−3 | 8 | 2.17 | 0.78 | 6.03 | 0.138 |
| EML5 | 44 | 41 | 0.89 | 0.58 | 1.36 | 0.581 | 9 | 1.40 | 0.67 | 2.91 | 0.366 | 17 | 0.83 | 0.47 | 1.47 | 0.519 |
| NTRK3 | 13 | 16 | 1.25 | 0.6 | 2.6 | 0.552 | 5 | 3.17 | 1.11 | 9.11 | 0.032 | 7 | 1.06 | 0.42 | 2.69 | 0.897 |
| MED23 | 39 | 53 | 1.29 | 0.85 | 1.96 | 0.225 | 10 | 1.69 | 0.83 | 3.43 | 0.148 | 26 | 1.35 | 0.82 | 2.25 | 0.242 |
| RNF175 | 22 | 37 | 1.64 | 0.97 | 2.79 | 0.066 | 4 | 1.40 | 0.47 | 4.12 | 0.545 | 19 | 2.00 | 1.07 | 3.75 | 0.031 |
| NCKAP1L | 13 | 23 | 1.69 | 0.86 | 3.35 | 0.130 | 1 | 0.53 | 0.07 | 4.09 | 0.542 | 14 | 2.16 | 1.01 | 4.65 | 0.048 |
| ABCC2 | 72 | 102 | 1.38 | 1.02 | 1.87 | 0.039 | 11 | 1.08 | 0.56 | 2.06 | 0.821 | 48 | 1.42 | 0.98 | 2.07 | 0.067 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dumont, M.; Weber-Lassalle, N.; Joly-Beauparlant, C.; Ernst, C.; Droit, A.; Feng, B.-J.; Dubois, S.; Collin-Deschesnes, A.-C.; Soucy, P.; Vallée, M.; et al. Uncovering the Contribution of Moderate-Penetrance Susceptibility Genes to Breast Cancer by Whole-Exome Sequencing and Targeted Enrichment Sequencing of Candidate Genes in Women of European Ancestry. Cancers 2022, 14, 3363. https://doi.org/10.3390/cancers14143363
Dumont M, Weber-Lassalle N, Joly-Beauparlant C, Ernst C, Droit A, Feng B-J, Dubois S, Collin-Deschesnes A-C, Soucy P, Vallée M, et al. Uncovering the Contribution of Moderate-Penetrance Susceptibility Genes to Breast Cancer by Whole-Exome Sequencing and Targeted Enrichment Sequencing of Candidate Genes in Women of European Ancestry. Cancers. 2022; 14(14):3363. https://doi.org/10.3390/cancers14143363
Chicago/Turabian StyleDumont, Martine, Nana Weber-Lassalle, Charles Joly-Beauparlant, Corinna Ernst, Arnaud Droit, Bing-Jian Feng, Stéphane Dubois, Annie-Claude Collin-Deschesnes, Penny Soucy, Maxime Vallée, and et al. 2022. "Uncovering the Contribution of Moderate-Penetrance Susceptibility Genes to Breast Cancer by Whole-Exome Sequencing and Targeted Enrichment Sequencing of Candidate Genes in Women of European Ancestry" Cancers 14, no. 14: 3363. https://doi.org/10.3390/cancers14143363