
Analysis of Corynebacterium silvaticum genomes from Portugal reveals a single cluster and a clade suggested to produce diphtheria toxin
- Published
- Accepted
- Received
- Academic Editor
- Joseph Gillespie
- Subject Areas
- Bioinformatics, Genomics, Microbiology, Toxicology, Veterinary Medicine
- Keywords
- Corynebacterium silvaticum, Pathogen, Diphtheria
- Copyright
- © 2023 Viana et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2023. Analysis of Corynebacterium silvaticum genomes from Portugal reveals a single cluster and a clade suggested to produce diphtheria toxin. PeerJ 11:e14895 https://doi.org/10.7717/peerj.14895
Abstract
Background
Corynebacterium silvaticum is a pathogenic, gram-positive bacterial species that causes caseous lymphadenitis in wild boars, domestic pigs and roe deer in Western Europe. It can affect animal production and cause zoonosis. Genome analysis has suggested that one strain from Portugal and one from Austria could probably produce the diphtheria toxin (DT), which inhibits protein synthesis and can cause death.
Methods
To further investigate the species genetic diversity and probable production of DT by Portuguese strains, eight isolates from this country were sequenced and compared to 38 public ones.
Results
Strains from Portugal are monophyletic, nearly identical, form a unique cluster and have 27 out of 36 known Corynebacterium virulence or niche factors. All of them lack a frameshift in the tox gene and were suggested to produce DT. A phylogenetic analysis shows that the species has diverged into two clades. Clade 1 is composed of strains that were suggested to have the ability to produce DT, represented by the monophyletic strains from Portugal and strain 05-13 from Austria. Clade 2 is composed of strains unable to produce DT due to a frameshifted tox gene. The second clade is represented by strains from Austria, Germany and Switzerland. Ten genome clusters were detected, in which strains from Germany are the most diverse. Strains from Portugal belong to an exclusive cluster. The pangenome has 2,961 proteins and is nearly closed (α = 0.968). Exclusive genes shared by clusters 1 and 2, and Portuguese strains are probably not related to disease manifestation as they share the same host but could play a role in their extra-host environmental adaptation. These results show the potential of the species to cause zoonosis, possibly diphtheria. The identified clusters, exclusively shaded genes, and exclusive STs identified in Portugal could be applied in the identification and epidemiology of the species.
Introduction
Corynebacterium silvaticum is a species of recently described gram-positive pathogenic bacteria (Dangel et al., 2020) that has been isolated from wild board and roe deer in Germany (Dangel et al., 2020; Möller et al., 2020a), Austria (JABGCO01), Switzerland (JAEANX01), and from domestic pigs in Portugal (Oliveira et al., 2014; Viana et al., 2020). The infection manifests in a disease similar to caseous lymphadenitis (CL) (Dangel et al., 2020), which is caused by C. pseudotuberculosis in goats and sheep (Dorella et al., 2006). Prior to an analysis and designation of a new species, strains from this species were identified as C. pseudotuberculosis or C. ulcerans (Oliveira et al., 2014; Dangel et al., 2019; Rau et al., 2019).
C. silvaticum is part of a group of six phylogenetically related pathogenic species that include C. diphtheriae, C. belfantii, C. rouxii, C. ulcerans, and C. pseudotuberculosis, which can produce diphtheria toxin (DT) when lysogenized by tox+ corynephages (Bernard & Funke, 2015; Dangel et al., 2020; Badell et al., 2020). This toxin causes cell death by inactivating protein synthesis (Murphy, 2011). One of the characteristics of C. silvaticum is that it is non-toxigenic, yet it has the tox gene (NTTB) (Dangel et al., 2020), caused by a frameshift in tox (Viana et al., 2020). The Portuguese strain PO100/5 and Austrian strain 05-13 do not have the characteristic frameshift in the tox gene caused by the insertion of two guanines (Möller et al., 2020a; Viana et al., 2020), suggesting that those strains are producers of DT. The isolation from domestic pigs suggests a potential for zoonotic transmission (Viana et al., 2020). Besides production of DT, cytotoxicity in human epithelial cells has recently been demonstrated (Möller et al., 2021).
The possibility of zoonotic transmission and the impact it could have on animal production implicate C. silvaticum as a potential threat to human health. In this work, we investigated the genetic diversity of the species by sequencing eight genomes from Portugal, with the aim of identifying genomic features that could be used for its control.
Materials & Methods
Genome assembly and taxonomy
The eight C. silvaticum strains from domestic pigs in Portugal used in the analysis were isolated by Oliveira et al. (2014) (Data S1). At the time the strains were classified as C. pseudotuberculosis. The genomes were sequenced using Illumina HiSeq 2500 (Illumina, San Diego, CA, USA) with 2 × 150 bp paired-end libraries. The quality of the sequencing reads was assessed by FastQC v0.11.9 (Andrews, 2015). Each genome was assembled using both reference-based and de novo assemblies. For the reference-based assembly, we used as reference the first version of C. silvaticum PO100/5 genome (CP021417.1, BV-BRC 65058.108). The tool used for read mapping and extraction of consensus sequence was UGENE 39 (Okonechnikov et al., 2012), with the plugins Bowtie v2.4.2 (Langmead & Salzberg, 2012) and SAMtools v0.1.19 (Li et al., 2009). For the de novo assemblies, we performed three assemblies using SPAdes v3.15.3 (Bankevich et al., 2012), Unicycler v0.4.8 (Wick et al., 2017) and Edena v3.131028 (Hernandez et al., 2008). Before finishing the assembly, the taxonomy of the sample was determined using the type strain genome server (Meier-Kolthoff & Göker, 2019). Then, the best de novo assembly was determined by QUAST v5.1.0rc1 (Gurevich et al., 2013). This assembly was scaffolded using CONTIGuator v2 (Galardini et al., 2011) using CP021417.1 as reference. The beginning of the chromosome was moved to the dnaA gene using the script moveDNAA.py (https://github.com/dcbmariano/scripts/blob/master/moveDNAA.py). The gaps were automatically closed using the contigs of the other three assemblies using GFinisher v1.4 (Guizelini et al., 2016) with CP021417.1 as reference. The assembly completeness and contamination were evaluated using CheckM 2 (https://github.com/chklovski/CheckM2).
For comparison of tox+ prophages, we reassembled the public genomes of strains 05-13 ( SRR11485666) and KL0182T (SRR7825394) (Data S1). The raw sequencing data was retrieved using fastq-dump from SRA Toolkit (https://github.com/ncbi/sratoolkit) and the assembly was performed with the method used in the strains from Portugal but replacing Edena’s assembly for the respective public assembly.
Clustering, typing and annotation
Genome clusters were determined using PopPUNK v2.6.0 (Lees et al., 2019) and the network was visualized using Cytoscape v3.9.1 (Shannon et al., 2003). The sequence type (ST) of the strains was determined using MLST v 2.0.4 (Larsen et al., 2012). The genomes were annotated using the Rapid Annotation using Subsystems Technology (RASTtk) pipeline (Brettin et al., 2015), implemented in the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) (Olson et al., 2022), and submitted to GenBank (Benson et al., 2012).
Characterization of C. silvaticum genomes from Portugal
Plasmids, insertion sequences and prophages were predicted using PlasmidFinder v2.1 (Carattoli et al., 2014), ISEScan v1.7.2.3 (Xie & Tang, 2017) and PHASTER (Arndt et al., 2016), respectively. Genomics islands were predicted for strain PO100/5 using GIPSy v1.1.3 (Soares et al., 2016), with C. glutamicum ATCC13032 (CP025533.1) as a non-pathogenic reference. CRISPR-Cas systems were identified using CRISPRCasFinder (Couvin et al., 2018). Virulence factors were predicted with Abricate v1.0.1 https://github.com/tseemann/abricate), with minimum identity and coverage values of 60%. Virulence and niche factors were identified using BV-BRC’s Proteome Comparison Tool, using a list of 37 genes described for C. silvaticum, C. ulcerans, C. pseudotuberculosis, C. diphtheriae, C. jeikeium and C. glutamicum (Trost et al., 2010; Trost et al., 2011; Tauch & Burkovski, 2015; Reardon-Robinson et al., 2015; Weerasekera et al., 2019; Möller et al., 2020b). A circular map of PO100/5 was generated using BRIG v0.95 (Alikhan et al., 2011).
The tox gene sequence was compared across Corynebacterium species to look for the frameshift described in C. silvaticum (Dangel et al., 2020). The representatives for C. silvaticum included the KL0182T and W25 strains from Germany, 5182 from Switzerland, 04-13 and 05-13 from Austria, and the eight strains from Portugal. Three other species were included in the comparison: C. ulcerans 0102 (AP012284.1), C. pseudotuberculosis 31 (CP003421.4) and C. diphtheriae NCTC13129 (NC_002935). The sequences were aligned using Jalview v2.11.1.4 (Waterhouse et al., 2009), with the MUSCLE algorithm (Edgar, 2004).
Comparative genomics with other strains
Thirty-eight public C. silvaticum genomes were obtained from BV-BRC and GenBank (Data S1) for a total of 46 when the Portuguese genomes were included. For samples available as sequencing reads (Data S1), we used the assemblies performed by Viana et al. (2020). A phylogenomic tree of C. silvaticum was built using BV-BRC’s Phylogenetic Tree Building tool, using the nucleotide and amino acid sequences from 1,000 shared genes. C. ulcerans NCTC 7910T was used as an external group. Average Nucleotide Identity (ANI) was calculated using FastANI v1.0 (Jain et al., 2018).
The distribution of orthologous gene groups across all genomes from all three species was estimated using OrthoFinder v2.5.4 (Emms & Kelly, 2019) and in-house scripts (Data S2). Here, the core genome is defined by orthogroups shared by all genomes, accessory genome is defined by orthogroups shared by more than one but not all genomes, and singletons are exclusive genes of a single genome. A pangenome is the entire repertoire of orthogroups found across all genomes. We used an in-house script to identify subsets of gene groups exclusively shared by (1) C. silvaticum strains from Portugal, (2) C. silvaticum strains from Portugal and strain 05-13 from Austria, (3) the remaining C. silvaticum strains.
The prophages of strains PO100/5, 05-13 and KL0182 were compared using tBLASTx v2.9.0+ or BLASTn v2.9.0+ (Camacho et al., 2009) and visualized using Artemis Comparison Tool (ACT) v18.1.0 (Carver et al., 2008; Carver et al., 2012). Possible misassemblies were investigated by mapping sequencing reads to the assembled genome or a reference using UGENE. The reassembled version and KL0182T was also used for genomic island prediction using GISPy to represent strains out of Portugal.
Results
Characterization of C. silvaticum genomes from Portugal
All strains from Portugal were identified as C. silvaticum (Data S3). The assemblies were estimated to be 99.9% complete with 0.19 or 0.2% contamination. No plasmids were detected. The genome sizes were ∼2.573 Mb, with 2,631 to 2,639 CDSs, 12 rRNA genes and 52 tRNA genes (Table 1). All genomes have three insertion sequences families (Table 1, Data S4), two complete and one to two incomplete prophages (Table 1, Data S5), and a Type I-E CRISPR-Cas system (Table 1, Data S6). The ANI values ranged from 99.9948 to 99.9998% (Data S7). The tox+ prophage was ∼38 Kb in all strains (Data S5). PO100/5 has 35 genomic islands in comparison to C. glutamicum ATCC13032 and one in comparison to KL0182T (Fig. 1, Data S8). KL0182T has 35 islands when compared to C. glutamicum (Data S8). Some islands are found in both PO100/5 and KL0182, and some are unique to each strain (Data S8). The new ST 795 was found in PO104/5, differing from the ST 578 by the new allele 65 of the gene atpA (Table 1, Data S9). Abricate identified virulence genes in four of the eight genomes (tox, relA, ideR and ureB) (Table 2, Data S10). The proteome comparison tool identified 28 out of 37 known Corynebacterium virulence or niche factors, although the pili genes spaCDEF were pseudogenized (Table 2, S11 File). All strains from Portugal and strain 05-13 from Austria have a tox gene that codes a DT with 560 amino acids with identical sequence (Data S12). The other strains have a frameshift that is caused by the insertion of two guanines in position 44 (Data S13). The frameshift results in a truncated protein of 17 amino acids (Fig. 2).
| Strain | PO25/4 | PO38/4 | PO39/4 | PO100/5 | PO101/5 | PO102/5 | PO104/5 | PO105/5 |
|---|---|---|---|---|---|---|---|---|
| Genbank accession | CP080461 | CP081182 | CP081179 | CP021417.2 | CP081180 | CP080459 | CP080460 | CP081181 |
| Completeness (%) | 99.9 | 99.9 | 99.9 | 99.9 | 99.9 | 99.9 | 99.9 | 99.9 |
| Contamination (%) | 0.2 | 0.19 | 0.2 | 0.19 | 0.19 | 0.19 | 0.2 | 0.2 |
| Size (bp) | 2,572,825 | 2,572,864 | 2,572,860 | 2,572,864 | 2,572,991 | 2,572,936 | 2,572,895 | 2,572,843 |
| Plasmid | – | – | – | – | – | – | – | – |
| CG content (%) | 54.40 | 54.40 | 54.40 | 54.40 | 54.40 | 54.40 | 54.40 | 54.40 |
| CDS | 2,631 | 2,633 | 2,636 | 2,633 | 2,636 | 2,635 | 2,634 | 2,639 |
| tRNA | 52 | 52 | 52 | 52 | 52 | 52 | 52 | 52 |
| rRNA | 12 | 12 | 12 | 12 | 12 | 12 | 12 | 12 |
| Repeat region | 24 | 24 | 24 | 24 | 24 | 24 | 24 | 24 |
| Sequence type | 578 | 578 | 578 | 709 | 578 | 709 | 795 | 578 |
| Insertion sequence | IS21, IS110 and IS256 | IS21, IS110 and IS256 | IS21, IS110 and IS256 | IS21, IS110 and IS256 | IS21, IS110 and IS256 | IS21, IS110 and IS256 | IS21, IS110 and IS256 | IS21, IS110 and IS256 |
| Prophages | 2 questionable, 2 incomplete | 2 questionable, 1 incomplete | 2 questionable, 2 incomplete | 2 questionable, 2 incomplete | 2 questionable, 1 incomplete | 2 questionable, 2 incomplete | 2 questionable, 2 incomplete | 2 questionable, 2 incomplete |
| CRISPR-Cas system | Type I-E | Type I-E | Type I-E | Type I-E | Type I-E | Type I-E | Type I-E | Type I-E |
Comparative genomics with other strains
The strains were represented by 10 clusters, with strains from Germany in seven of them, being the most diverse. All strains from Portugal were in an exclusive cluster. Within strains from Austria, 05-13 had its own cluster, while 04-13 is part of a bigger cluster that includes strains from Germany and the single one from Switzerland (Fig. 3). The phylogenetic tree shows two clades. Clade 1 is monophyletic and has the strains from Portugal and strain 05-13 from Austria. Clade 2 has the remaining strains from Austria, Germany, and Switzerland (Fig. 4). The ANI values ranged from 99.7539 to 100% for all strains, and 99.9632 to 100% within the same clade. The difference between clades ranged from 0.2461 to 0.1729% (Data S7).
The alignment of the tox+ prophages of PO100/5, 05-13 (Clade 1) and KL0182T (Clade 2) shows nearly identical sequences for PO100/5 and 05-13, with a size of ∼38 Kb containing an additional 5.8 Kb sequence upstream the tox gene in comparison to KL0182T (Wild boar, Germany, ∼32.8 Kb) (Fig. 5). The additional 5.8 Kb is a repeat and is also present in KL0182T but in GI 18 rather than in tox+ prophage (Fig. 6). Mapping the sequencing reads of KL0182T to its assembled genome and to PO100/5 genome confirmed that part of the prophage sequence is in another region of the genome (Data S14). Besides the insertion, coding sequences can be fragmented or fused when the tox+ prophage from PO100/5 and KL0182T are compared (Fig. 5).
Across the 46 genomes, the pangenome, core genome, shared genome and singletons were 2,961, 2,227 (75.21%), 623 (21.04%) and 111 (3.75%) orthogroups, respectively (Data S15). The value of α was 0.968 (Data S16). The subsets of exclusively shared orthogroups were: 19 in C. silvaticum strains from Portugal, 25 in C. silvaticum strains from Portugal and strain 05-13 from Austria, and 36 from the remaining strains (Data S17 and S18). Of the 28 Corynebacterium virulence/niche factors described in the literature and identified in strains from Portugal (Table 2, S11 File), 19 are part of the core genome and eight are part of the shared genome (Data S15).
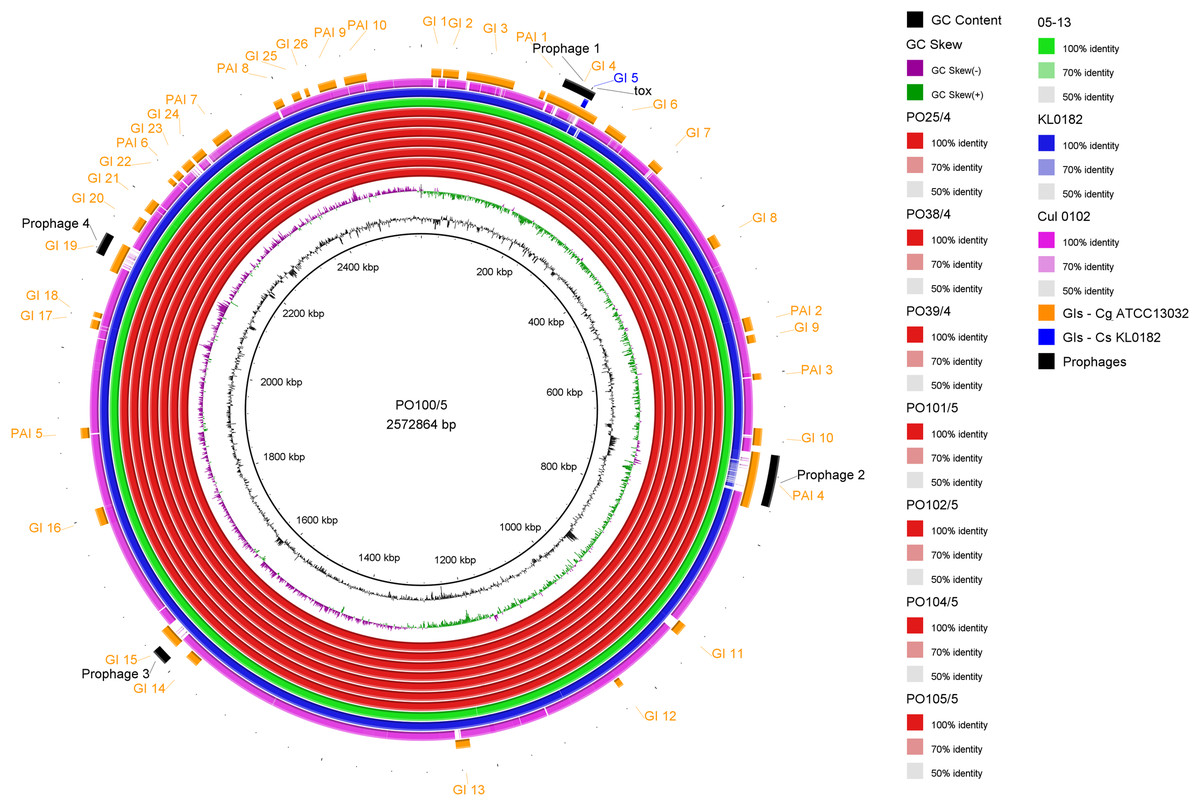
Figure 1: Circular map of Corynebacterium silvaticum PO100/5 genome generated using BRIG v0.95.
Genomic islands and prophage detection were performed using GIPSy and PHASTER, respectively. Cul –C. ulcerans, GIs –genomic islands, Cg –C. glutamicum, Cs –C. silvaticum.{kind=link}
Discussion
Strains from Portugal and production of diphtheria toxin
All the Portuguese strains are monophyletic (Fig. 4), from the same cluster (Fig. 3), and nearly identical (Data S7), suggesting that they were derived from a single clone. The fact that the Portuguese strains have a most recent common ancestor with Austrian strain 05-13 suggests that the initial infection originated in Austria.
Eight virulence and niche factors identified in strains from Portugal are in the accessory genome (Data S14). spaDEF and srtC are part of a pilus cluster and are fragmented in different genomes. The absence of the tox gene in some strains is certainly an assembly artifact, since the strains lacking it were shown to have it (Dangel et al., 2019). The absence of sialidase (nanH) and serine protease (cpfrc_00397) in some strains could also be a sequence or assembly artifact, since they were detected in 45 out of 46 strains. The venom serine protease (vsp1) was found in all strains from Portugal and KL1008 from Germany, but the advantages for host colonization in comparison to other strains that lack the protein must be elucidated. Among the others, the niche factors Trypsin-like serine protease (Uniprot A0A5C5F2T7), cwlH (A0A5C5F4U0) and rfpI (A0A5F0A739) are part of the core genome (Data S15) and shared by all of the strains examined. These three proteins have been shown to be the most abundant extracellular proteins produced in vitro by C. silvaticum W25, representing 88.1, 2.2 and 1.3%, respectively (Möller et al., 2020b).
| Type | Gene | Product | Presence | Reference locus tag | Reference species | Reference |
|---|---|---|---|---|---|---|
| Niche | – | C. diphtheriae DIP0733 homolog | Yes | CULC22_00609 | Cul | Tauch & Burkovski (2015) |
| Niche | – | Secreted subtilisin-like serine protease | Yes | cpfrc_00397 | Cp | Trost et al. (2010) |
| Niche | – | Secreted subtilisin-like serine protease | Yes, except in PO25/4 | cpfrc_01634 | Cp | Trost et al. (2010) |
| Niche | – | Secreted trypsin-like serine protease | No | cpfrc_00562 | Cp | Trost et al. (2010) |
| Niche | – | Secreted SGNH-hydrolase | Yes | cpfrc_00536 | Cp | Trost et al. (2010) |
| Niche | – | Trypsin-like serine protease | Yes | FIT55_05760, A0A5C5F2T7 | Cs | Möller et al. (2020b) |
| Niche | accD3 | Acyl-CoA carboxylase b-subunit involved in mycolic acid synthesis | Yes | cpfrc_01953 | Cp | Trost et al. (2010) |
| Niche | asa | Ceramidase | No | jk1103 | Cj | Tauch et al. (2005), Tauch & Burkovski (2015) |
| Niche | che | Cholesterol esterase | No | jk2054 | Cj | Tauch et al. (2005), Tauch & Burkovski (2015) |
| Niche | choE | Cholesterol oxidase | No | jk0629 | Cj | Tauch et al. (2005), Tauch & Burkovski (2015) |
| Niche | cwlH | Cell wall-associated hydrolase | Yes | CULC809_01521 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | dtsR1 | Acetyl-CoA carboxylase b-subunit involved in fatty acid synthesis | Yes | cpfrc_00492 | Cp | Trost et al. (2010) |
| Niche | dtsR2 | Acyl-CoA carboxylase b-subunit involved in mycolic acid synthesis | Yes | cpfrc_00491 | Cp | Trost et al. (2010) |
| Niche | endoE | Endoglycosidase E (former corynebacterial protease CP40) | Yes | CULC809_01974 | Cul | Trost et al. (2011) |
| Niche | mdbA | Thiol-disulfide oxidoreductase | Yes | DIP1880 | Cd | Reardon-Robinson et al. (2015), Tauch & Burkovski (2015) |
| Niche | nanH | Sialidase (neuraminidase H) | Yes | CULC809_00434 | Cul | Trost et al. (2011) |
| Niche | nor | Nitric oxide reductase | No | cpfrc_00128 | Cp | Trost et al. (2010) |
| Niche | nrpS1 | Nonribosomal peptide synthetase 1 | Yes | cpfrc_00565 | Cp | Trost et al. (2010) |
| Niche | nrpS2 | Nonribosomal peptide synthetase 2 | No | cpfrc_00180 | Cp | Trost et al. (2010) |
| Niche | rhuM | RhuM-like protein | No | CulFRC58_0285 | Cul | Trost et al. (2011); Weerasekera et al. (2019) |
| Niche | rpfA | Resuscitation-promoting factor A (muralytic enzyme) | Yes | cpfrc_00594 | Cp | Trost et al. (2010) |
| Niche | rpfB | Resuscitation-promoting factor B (muralytic enzyme) | Yes | cpfrc_00679 | Cp | Trost et al. (2010) |
| Niche | rpfI | Resuscitation-promoting factor-interacting protein | Yes | CULC809_01133 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | spaB | Surface-anchored protein (minor pilus subunit) | Yes | CULC809_01980 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | spaC | Surface-anchored protein (tip pilus protein) | Pseudogene, no CWSS | CULC809_01979 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | spaD | Surface-anchored protein (major pilus subunit) | Pseudogene, no CWSS | CULC809_01952 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | spaE | Surface-anchored protein (minor pilus subunit) | Pseudogene, no SP | CULC809_01950 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | spaF | Surface-anchored protein (tip pilus protein) | Pseudogene, no SP | CULC809_01949 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | srtA | Sortase A | Yes | CULC809_01981 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | srtB | Sortase B | Yes | CULC809_01953 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | srtC | Sortase C | Yes | CULC809_01951 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Niche | tspA | Trypsin-like serine protease | No | CULC809_01848 | Cul | Trost et al. (2011) |
| Niche | vsp1 | Venom serine protease | Yes | CULC809_00509 | Cul | Trost et al. (2011) |
| Niche | vsp2 | Venom serine protease | Yes | CULC809_01964 | Cul | Trost et al. (2011) |
| Virulence | pld | Phospholipase D | Yes | ET810_03855 | Cs | Trost et al. (2011); Tauch & Burkovski (2015) |
| Virulence | rbp | Shiga-like ribosome-binding protein | No | CULC809_00177 | Cul | Trost et al. (2011); Tauch & Burkovski (2015) |
| Virulence | tox | Diphtheria toxin | Yes | DIP0222 | Cd | Tauch & Burkovski (2015) |
| Virulence | relA | Guanosine-3′, 5′-bis(diphosphate) 3′-pyrophosphohydrolase / GTP pyrophosphokinase, (p)ppGpp synthetase II | Yes | – | – | Abricate |
| Virulence | ideR | Iron-dependent repressor IdeR/DtxR | Yes | – | – | Abricate |
| Virulence | ureB | Urease alpha subunit | Yes | – | – | Abricate |
| Resistance | rpoB2 | Rifampin-resistant beta-subunit of RNA polymerase | Yes | – | – | Abricate |
| Resistance | mtrA | Two component system response regulator MtrA | Yes | – | – | Abricate |
| Resistance | rbpA | RNA-polymerase binding protein which confers resistance to rifampin | Yes | – | – | Abricate |
Notes:
- CWSS
-
cell wall sorting signal
- SP
-
signal peptide
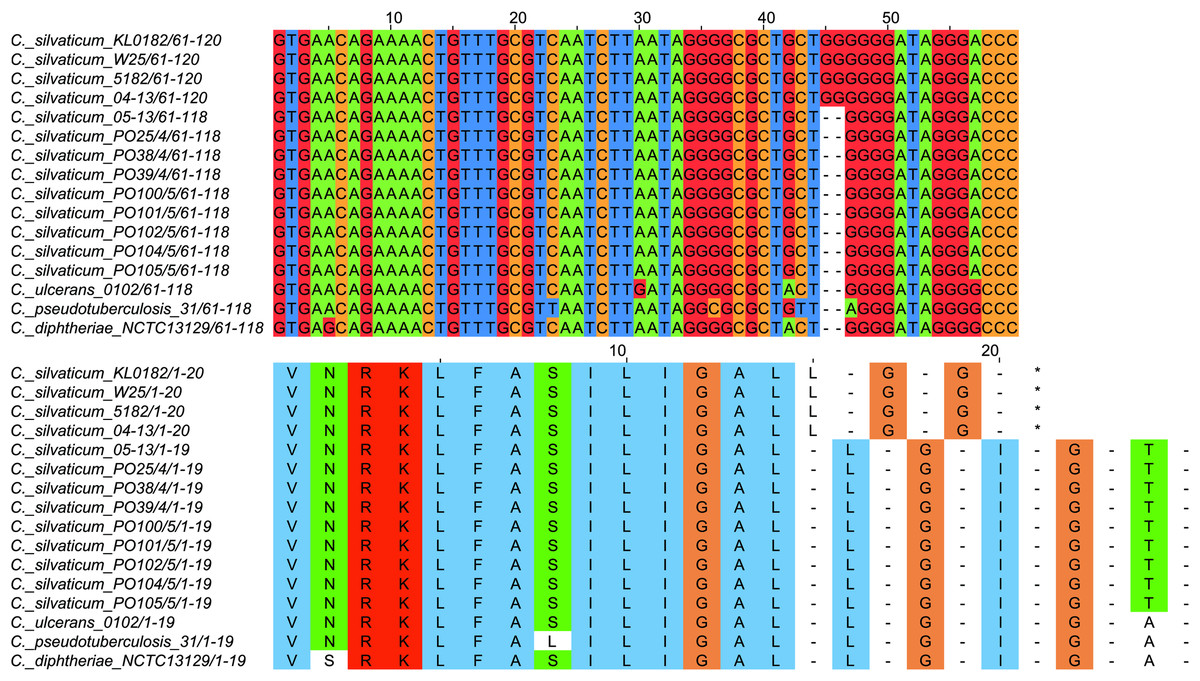
Figure 2: Alignment of the tox gene’s first 60 nucleotides and respective amino acids of strains from Corynebacterium silvaticum, C. ulcerans, C. pseudotuberculosis and C. diphtheriae.
C. silvaticum strains from Portugal PO25/4, PO38/4, PO39/4, PO100/5, PO101/5, PO102/5, PO104/5 and PO105/5, and the strain 05-13 from Switzerland do not have the insertion of two guanines that led to a frameshift in other strains from this species. The frameshift results in a truncated protein with 17 amino acids. The alignment was performed using MUSCLE algorithm implemented in Jalview v2.11.1.4.{kind=link}
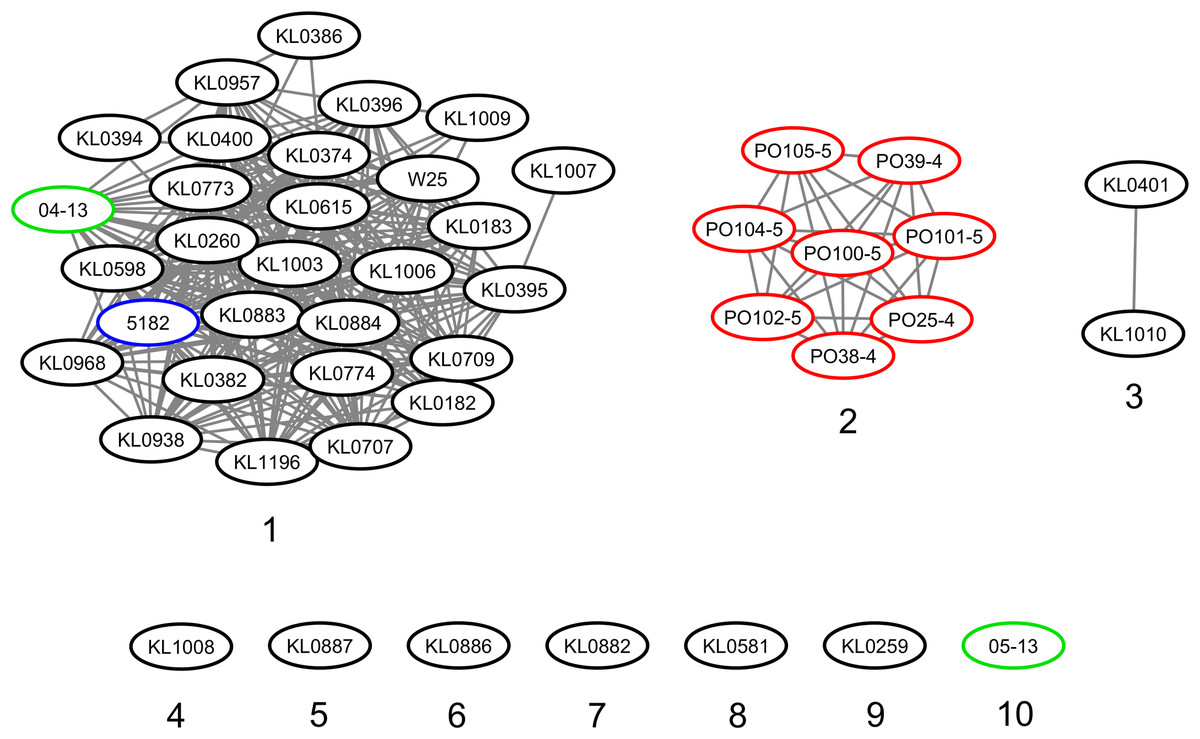
Figure 3: Clusters of Corynebacterium silvaticum genomes.
Countries of sample isolation: Austria (green), Germany (black), Portugal (red), Switzerland (blue).{kind=link}

Figure 4: Phylogenomic tree of Corynebacteriumsilvaticum strains.
The phylogeny was inferred using the Phylogenetic Tree service in PATRIC, which uses RAxML with 100 rounds of “rapid bootstrapping”, and the codon sequences from 1,000 single-copy shared genes. Bootstrap values are represented by a color scale from red (70%) to green (100%). Corynebacterium ulcerans NCTC7910 was used as an outgroup.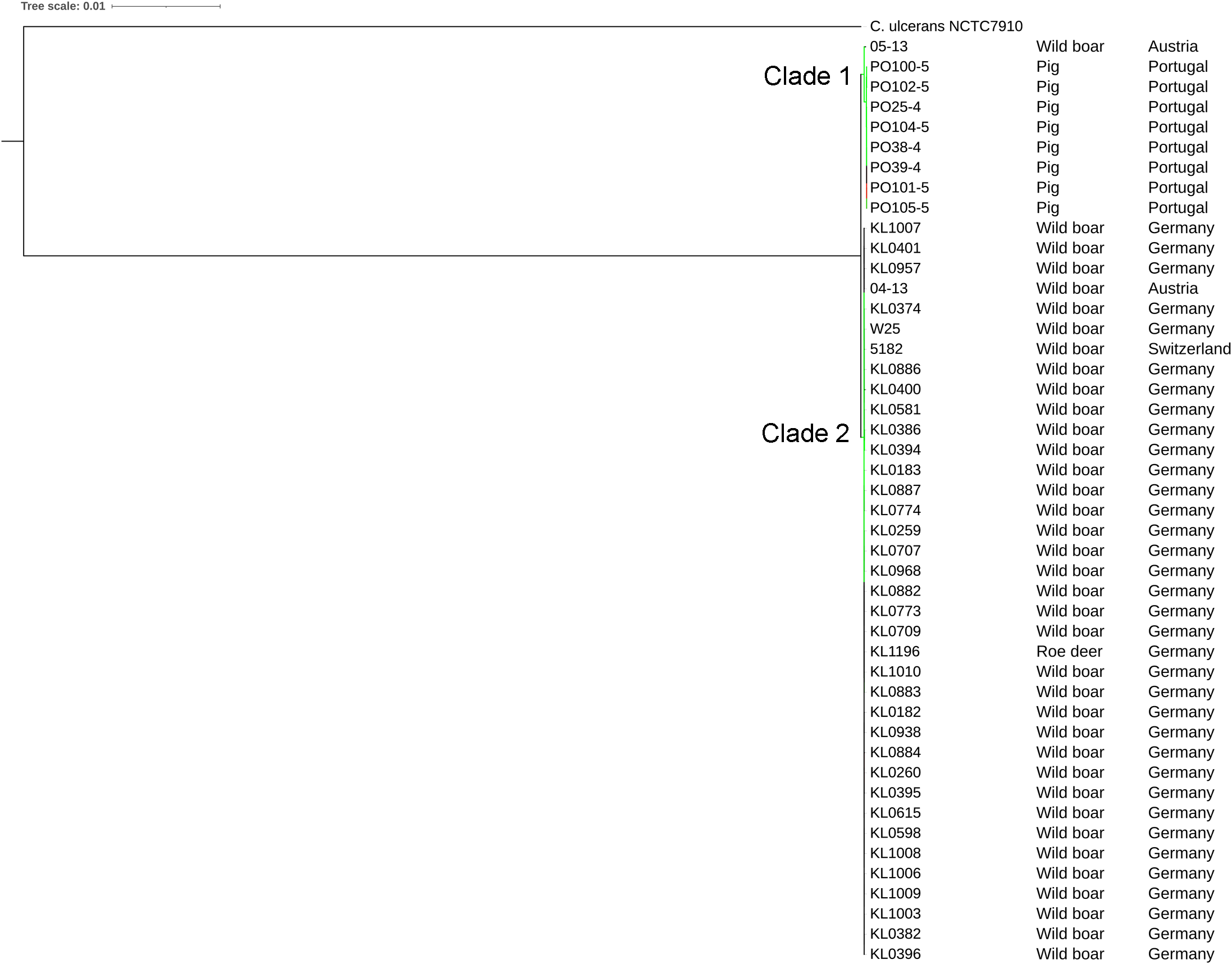{kind=link}
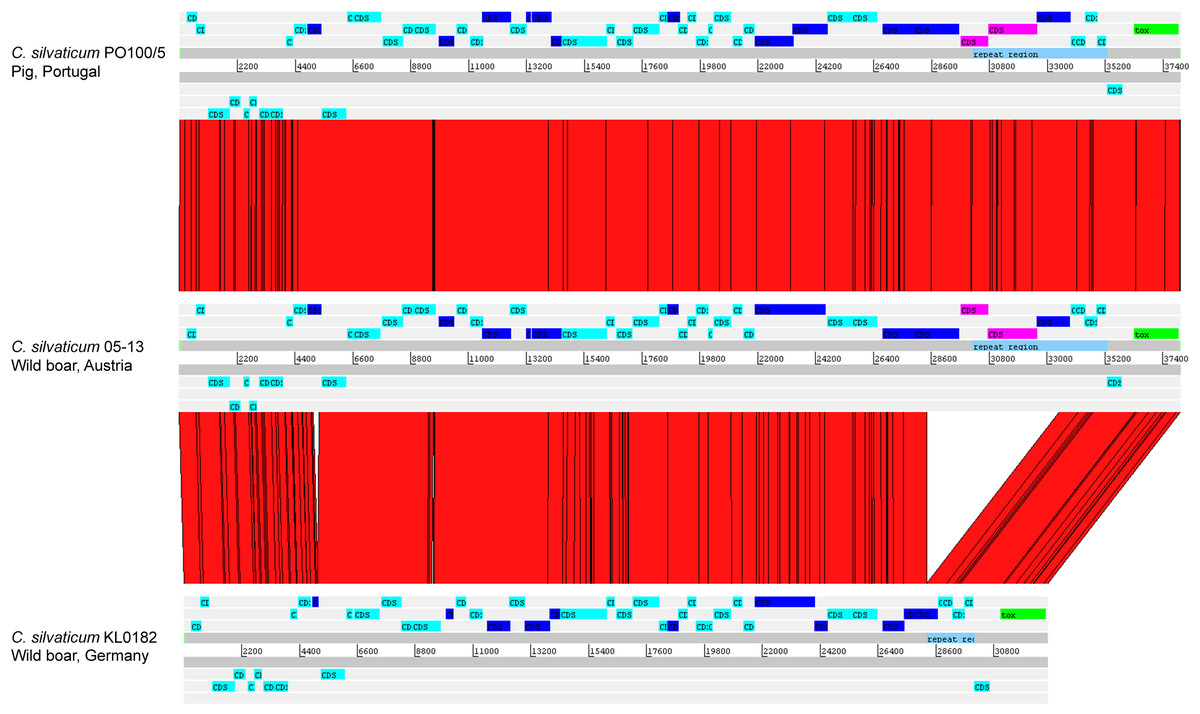
Figure 5: Alignment of tox+ prophages from Corynebacterium silvaticum strains.
Coding sequences in dark blue are fragmented or fused in at least one genome. Coding sequences in pink are not present in strain KL0182T prophage.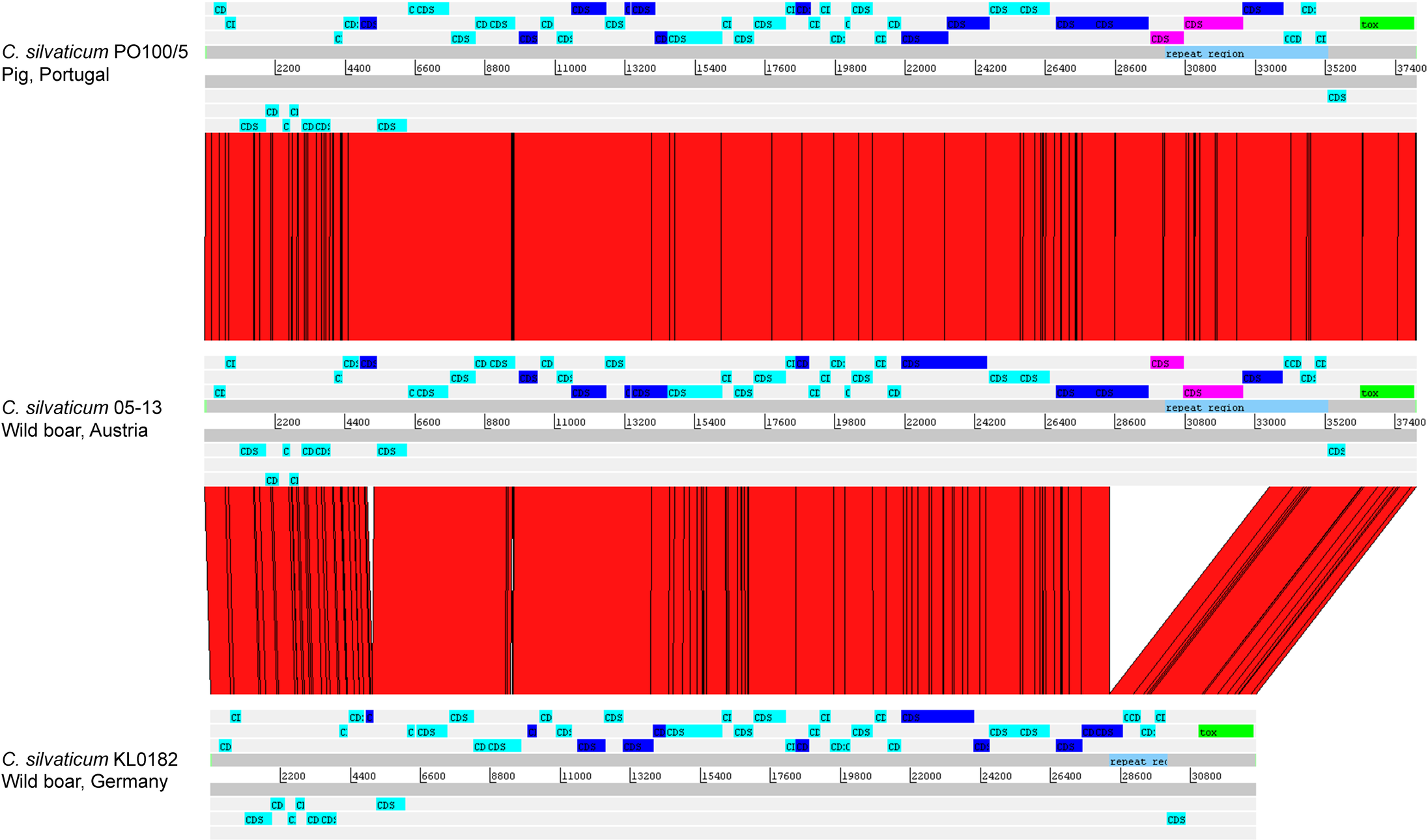{kind=link}
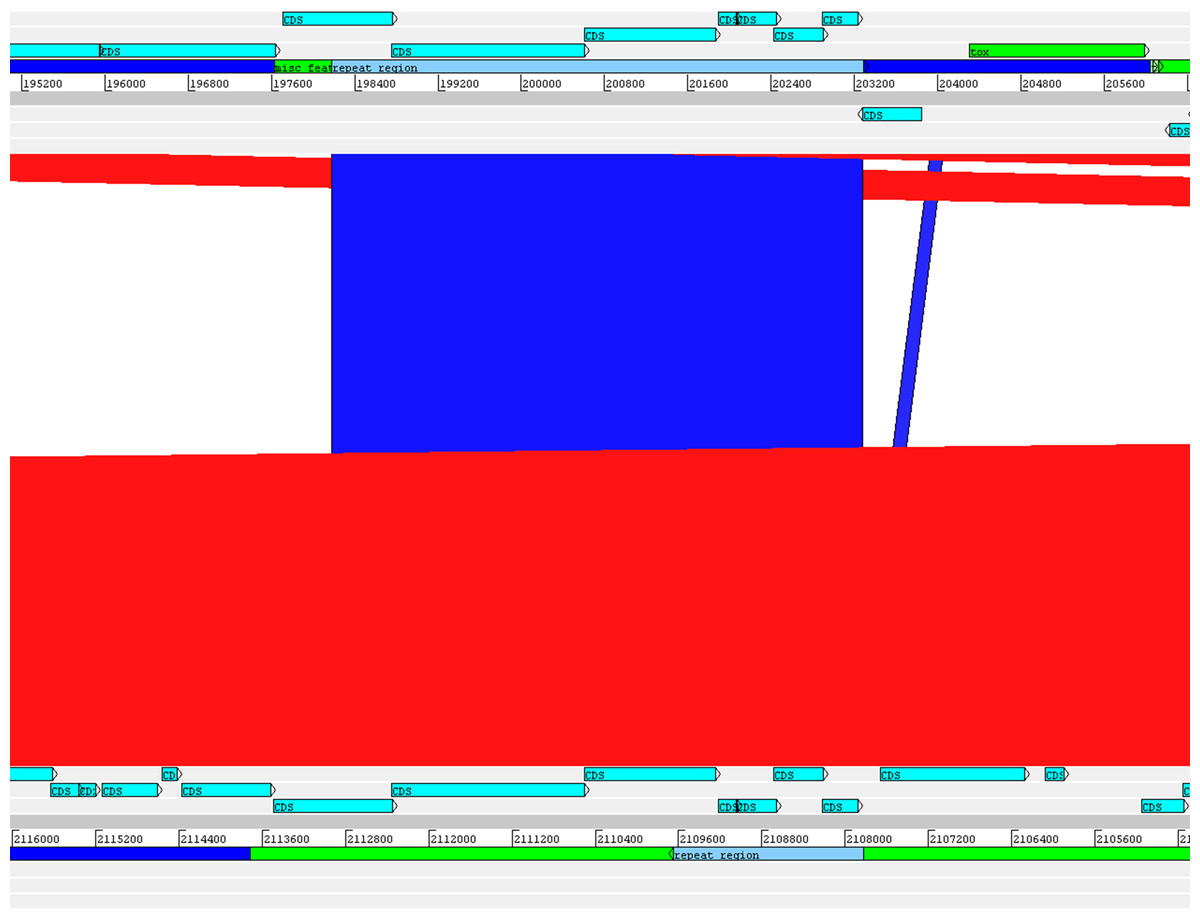
Figure 6: Alignment of part of the tox+ prophage from Corynebacterium silvaticum PO100/5 (top) and KL0182T (bottom).
The absent sequence in strain KL0182T tox+ prophage is part of a repeat region and is in another part of the genome , inside genomic island GI 18 (bottom, green). Dark blue highlight in chromosome –prophage, green highlight in chromosome –genomic island.{kind=link}
C. silvaticum was described as non-toxigenic tox gene-bearing (Dangel et al., 2020). An insertion of two guanines in tox caused a frameshift and the pseudogenization of this gene (Möller et al., 2020a). However, the strains PO100/5 from Portugal and 05-13 lack the insertion and therefore could produce the toxin (Viana et al., 2020). We confirmed that all eight strains from Portugal lack that insertion that knocks out this gene (Fig. 2, Data S13) and are probable DT producers. The production of DT in strains 4-13 and 05-13 from Austria was inferred by RT-qPCR (Schaeffer et al., 2020), targeting subunits A and B of the DT (Mothershed et al., 2002). However, strain 04-13 has the typical frameshift in tox. The primers binding sites start from position 312, while the frameshift occurs in position 44 (Fig. 2, Data S13), so the frameshift cannot not be detected. It was suggested that DT production must be confirmed on tox-positive isolates by an Elek test, due to the description of tox-positive and Elek-negative strains (Schuhegger et al., 2008; Berger et al., 2014). This shows a limitation of the current RT-qPCR for detection of DT, and that 04-13 probably does not produce the toxin.
DT is a virulence factor because it only works inside of host cells and damages them (Tauch & Burkovski, 2015), but that may not impact C. silvaticum’s host range in the wild. The species causes CLA in its known hosts (Dangel et al., 2020), a disease manifestation like the non-DT-producing C. pseudotuberculosis biovar ovis causes in goats and sheep (Dorella et al., 2006). CLA is related to the toxin Phospholipase D (pld) (Dorella et al., 2006) that is also produced by C. silvaticum (Dangel et al., 2020). Another possibility is that the DT is required only for the infection of as yet unidentified hosts. It has been hypothesized that the production of DT is required for C. pseudotuberculosis biovar equi to infect buffalo (Viana et al., 2017). If DT production is not required, this could relax the selective pressure to keep a functional tox gene and could lead to accumulation mutations that would result in the loss of function (Figs. 2 and 5) seen in some populations of Germany, Austria and Switzerland.
Genome diversity of C. silvaticum
The phylogenetic tree showed that strains from Portugal and 05-13 from Austria formed Clade 1 and the remaining strains formed Clade 2 (Fig. 4). The two strains from Austria (04-13 and 05-13) are in different clades, suggesting different geographical origins. The strains from Portugal are in the same clade and nearly identical to 05-13, which suggests an Austrian origin.
Although the genomes had high ANI values, of at least 99.7% (Data S7), we could identify 10 clusters (Figs. 3 and 4). Strains from Germany had higher diversity, with strains in seven clusters, probably due to the higher number of samples and isolation from wild animals. All strains from Portugal had an exclusive cluster. As they were isolated from domestic pigs in two farms (Oliveira et al., 2014) this represents the spread of only one clone. More strains must be analyzed to assess the diversity of the species.
Across the 46 genomes, we identified a pangenome of 2,961 orthogroups, core genome of 2,227, 623 shared orthogroups, 111 singletons and an α of 0.968 (Data S5 and Data S15). The pangenome is nearly closed, which agrees with the high ANI values between the genomes. We identified exclusively shared orthogroups in strains from Portugal (19 orthogroups), Clade 1 (27 orthogroups), and Clade 2 (36 orthogroups) (Data S17 and S18). Those genes are probably not involved in the manifestation of the disease as it is the same manifestation (CL) in the known hosts but could be required for survival outside of the host.
With the presence of C. silvaticum in wild and domestic animals, cytotoxicity to human epithelial cells (Möller et al., 2021) and the probable production of DT, this species has the potential to cause zoonosis and diphtheria. Human transmission could occur via occupational exposure, as seen in C. pseudotuberculosis (Dorella et al., 2006). In this context, we identified 10 clusters (Fig. 3), exclusively shared genes of clades and Portuguese strains (Data S17 and S18), and the exclusive STs from Portugal (Data S9). This information can be applied to the identification and epidemiology of C. silvaticum.
Conclusions
In C. silvaticum, Clade 1 includes strains that have the potential to produce DT, which is missing in Clade 2 (non-DT-producing). Both clades can be identified by genes that, while probably not important for their interactions within the host environment, could play a role in survival in the environment. Portuguese strains are monophyletic, nearly identical, form a unique cluster and probably produce DT. We showed that the species has the potential to cause zoonosis and diphtheria, and genome clusters, STs and exclusive genes that can be applied to its epidemiology.