The Cell-Penetrating Peptide Tat Facilitates Effective Internalization of PSD-95 Inhibitors Into Blood–Brain Barrier Endothelial Cells but less Efficient Permeation Across the Blood–Brain Barrier In Vitro and In Vivo
 Emma Lisa Al Humaidan1†
Emma Lisa Al Humaidan1†  Sidse Lund Pedersen1†
Sidse Lund Pedersen1†  Annette Burkhart2
Annette Burkhart2  Charlotte Laurfelt Munch Rasmussen2
Charlotte Laurfelt Munch Rasmussen2  Torben Moos2
Torben Moos2  Peter Fuchs1 Eduardo Filipe Alves Fernandes3
Peter Fuchs1 Eduardo Filipe Alves Fernandes3  Burak Ozgür1 Kristian Strømgaard3 Anders Bach3 Birger Brodin1
Burak Ozgür1 Kristian Strømgaard3 Anders Bach3 Birger Brodin1  Mie Kristensen1*
Mie Kristensen1*- 1Department of Pharmacy, University of Copenhagen, Copenhagen, Denmark
- 2Neurobiological Research and Drug Delivery, Aalborg University, Aalborg, Denmark
- 3Department of Health Science and Technology, Aalborg University, Aalborg, Denmark
Inhibition of the interaction between the scaffolding protein PSD-95 and the NMDA receptor has been shown to obstruct ischemic stroke-triggered excitotoxic reactions, leading to neuronal death. The peptides NR2B9c and N-dimer are inhibitors of this interaction. Delivery of the peptides to the brain is challenging due to the general low blood–brain barrier (BBB) permeability. NR2B9c and N-dimer have therefore been conjugated to the cell-penetrating peptide (CPP) Tat, to facilitate blood–brain barrier permeation. However, the BBB permeation of Tat-NR2B9c and Tat-N-dimer has not been fully elucidated. We recently demonstrated that the BBB permeation in vitro and in vivo was lowered upon conjugation of NR2B9c or N-dimer to Tat. In the present study, we aimed to further understand the impact of cargo conjugation to Tat with respect to interaction with and permeation across the BBB in vitro and in vivo. The peptides were labeled with the fluorophore TAMRA (T) and demonstrated efficient Tat-mediated uptake into BBB endothelial cells but differed in their degree of plasma membrane interaction and embedding (T-Tat-NR2B9c = T-Tat > T-Tat-N-dimer) as well as in their chemical stability (T-Tat-N-dimer = T-Tat > T-Tat-NR2B9c). The Tat conjugates all displayed a similar degree of self-association and/or plasma protein adsorption. T-Tat-NR2B9c and T-Tat affected the BBB integrity but not the permeation of the paracellular marker C14-mannitol. T-Tat-NR2B9c and T-Tat-N-dimer displayed less efficient permeation across an in vitro model representing the healthy BBB, when compared to T-Tat, and low BBB permeation in healthy rats.
Introduction
Ischemic stroke is a leading cause of death. Current therapy is limited to systemic administration of a thrombolytic agent (Alteplase) and thrombectomy (Catanese et al., 2017), which, however, do not stop the excitotoxic reactions, leading to neuronal death and long-term disabilities. At present, pharmacological treatment to relieve neuronal damage resulting from an ischemic stroke is non-existing. Inhibition of the interaction between the neuronal scaffolding protein PSD-95 and the NMDA receptor may prevent neuronal damage after ischemic stroke (Ballarin and Tymianski, 2018; Ugalde-triviño and Margarita, 2021). Aarts et al. and Bach et al. designed the peptides NR2B9c (Aarts et al., 2002) and N-dimer (Bach et al., 2012), respectively, which effectively impedes this interaction. The 11-mer Tat peptide (YGRKKRRQRRR) originates from the HIV transactivator of transcription protein (Vivès et al., 1997) and belongs to the group of cell-penetrating peptides (CPPs) (Copolovici et al., 2014). Tat has demonstrated excellent potential in translocating across the plasma membrane of various cell types, with or without a conjugated cargo molecule (Tünnemann et al., 2006; Cleal et al., 2013). In addition, an early study by Schwarze et al. pointed toward the ability of Tat to transport a conjugated protein cargo, not only into cells but also across the blood–brain barrier (BBB) (Schwarze et al., 1999). Aarts et al. and Bach et al. conjugated NR2B9c and N-dimer, respectively, to Tat, to facilitate permeation across the BBB and internalization into neurons for target engagement. Systemic delivery of Tat-NR2B9c (also named NA-1 or nerinetide) and Tat-N-dimer (also named AVLX-144) has demonstrated reduction in neuronal death and improved cognition in animals subjected to experimental stroke (Aarts et al., 2002; Bach et al., 2012; Cook et al., 2012; Sommer et al., 2016). Tat-NR2B9c completed a phase III clinical trial (ESCAPE NA-1) in 2020 (Hill et al., 2020; ClinicalTrials.gov, 2022a), whereas Tat-N-dimer successfully completed a phase I clinical trial in 2021 (ClinicalTrials.gov, 2022b). However, the ESCAPE NA-1 trial overall demonstrated no neuroprotective effect upon systemic delivery of Tat-NR2B9c after administration of the thrombolytic agent Alteplase (Hill et al., 2020). On the other hand, Tat-NR2B9c did exert its neuroprotective effect when administered before Alteplase. A protease-resistant analog of Tat-NR2B9c was recently developed to solve the poor plasma half-life of Tat-NR2B9c in the presence of Alteplase (Nunez et al., 2021). In contrast, Tat-N-dimer displays improved chemical stability (Bach et al., 2012; Kristensen et al., 2020b) and a 1,000-fold better target affinity (Bach et al., 2012), when compared to Tat-NR2B9c. We recently demonstrated that cargo conjugation to Tat affects BBB interaction and permeation and that to different extents when comparing Tat-NR2B9c and Tat-N-dimer (Kristensen et al., 2020b).
With this study, we aim to further understand the underlying mechanisms resulting in the observed differences in BBB permeation of Tat-NR2B9c and Tat-N-dimer in terms of peptide self-association and plasma protein adsorption as well as their effect on the BBB integrity. The peptides were labeled with the fluorophore TAMRA (T) to allow sensitive quantification and visualization. We used a human stem cell–based in vitro BBB model to study the interaction between the peptides and the endothelial membrane, the BBB permeation, and the effects on the barrier integrity including monolayer morphology and tight junction protein expression. In vivo brain delivery was studied in rats along with an assessment of biodistribution to evaluate the off-target distribution.
We demonstrate effective uptake of T-Tat-NR2B9c and T-Tat-N-dimer into BBB endothelial cells in vitro but less efficient in vitro and in vivo permeation across the healthy BBB.
Materials and Methods
Materials
6-carboxytetramethylrhodamine [TAMRA (T)–conjugated NR2B9c (KLSSIESDV], Tat (YGRKKRRQRRR), and Tat-NR2B8c (YGRKKRRQRRRKLSSIESDV) were purchased as HCl salts with a purity >95% from Biomatik (Cambridge, ON, Canada). T-Tat-N-dimer (YGRKKRRQRRRNPEG4IETDV2) was custom-made as HCl salts with a purity >95% by Wuxi AppTec (Shanghai, China). The bEND3 cell line was obtained from American Type Culture Collection (Manassas, VA, United States). The BIONi010-C cell line was a kind gift from Bioneer A/S (Hørsholm, Denmark). HEPES {2-[4-(2-hydroxyethyl)-piperazin-1-yl] ethane sulfonic acid} was purchased from Applichem GmbH (Darmstadt, Germany). Hank’s balanced salt solution (HBSS) and Alexa Fluor® 488–conjugated phalloidin were purchased from Life Technologies (Taastrup, Denmark). Dulbecco’s Modified Eagle’s Medium (DMEM)–high glucose was purchased from PAA-Laboratories (Pasching, Austria). Fetal bovine serum (FBS) was obtained from Gibco and Fisher Scientific (Slangerup, Denmark). Penicillin and streptomycin were from Bio Whittaker Cambrex (Vallensbaek, Denmark). Matrigel® (354230) and mTeser1 were purchased from Stem Cell Technologies (Vancouver, BC, Canada), Versene and Accutase were obtained from Thermo Fisher Scientific (Waltham, MA, United States), and ROCK inhibitor Y27632 dihydrochloride was obtained from Tocris Bioscience (Bristol, Great Britain). Human basic fibroblast growth factor (bFGF) was purchased from R&D Systems (Minneapolis, MN, United States). DMEM/Nutrient Mixture F-12 Ham with l-glutamine and HEPES (DMEM/F12), Eagle’s minimum essential medium (MEM), nonessential amino acids, l-glutamine, β-mercaptoethanol, knockout replacement serum (KOSR), human endothelial serum-free media (hESFM), and bovine poor plasma-derived serum (PDS) were obtained from Thermo Fisher Scientific (Waltham, MA, United States). Paraformaldehyde (PFA) was purchased from Alfa Aesar (Thermo Fisher GmbH, Karlsruhe, Germany). Cell Proliferation Kit II (XTT) was purchased from Roche (Basel, Switzerland). Ultrapure water was collected from a PURELAB Flex 4 system (ELGA LabWater, High Wycombe, United Kingdom). Ethanol (96%) was purchased from VWR International (Radnor, PA, United States). All other chemicals were obtained from Merck Life Science (Søborg, Denmark), unless otherwise stated.
Peptide Self-Association and/or Plasma Protein Adsorption
T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer (10 µM) was incubated in buffer [10 mM HEPES HBSS supplemented with 0.05% bovine serum albumin (BSA) and adjusted to pH 7.4 (hHBSS)] or cell medium (hESFM supplemented with 1% PDS) in low-binding microtubes for 1 h at 37°C with orbital shaking (90 rpm) (UNIMAX 2010; Heidolph, Schwabach, Germany). The peptides were transferred to 30-kDa cutoff Microcon® centrifugal filters (Merck Millipore, Ireland) and subjected to centrifugation for 12 min at room temperature and 14,000 g using a Sigma 1–15K centrifuge (DJB Labcare, Newport Pagnell, England). Subsequently, the filter inserts were inverted before the addition of 100 µl of buffer or cell medium and centrifuged for 12 min at room temperature at 6,000 g. Non-associated peptide was obtained from the flow-through, and self-associated/plasma protein–adsorbed peptide was retained by the filter and recovered, subsequently. Non-associated and self-associated/plasma protein–adsorbed peptides were quantified using a NovoStar fluorescence plate reader (BMG Labtech, Offenburg, Germany) with excitation and emission wavelengths set to 542 and 568 nm, respectively.
Peptide Stability
T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer (50 µM) were incubated in cell medium or rat plasma for 3 h at 37°C with orbital shaking (90 rpm). Samples (20 µl) were withdrawn (0, 5, 10, 15, 30, 45, 90, 120, and 180 min) and 50 µl of 6 M urea to denature plasma proteins was added. Subsequently, 50 µl of 20% (w/v) trichloroacetic acid in acetone was added for protein precipitation and incubated for 10 min before centrifugation for 5 min at 5°C and 10,000 g. The acetone phase containing the peptide was diluted 1:1 with 0.1% trifluoroacetic acid and filtered (pore size of 0.22 µm) before UPLC analysis using a Waters Acquity system equipped with a BEH C18 reverse phase column (2.1 × 50 × 1.7 µm). A linear gradient of mobile phase A (94.9% ultrapure water, 5% acetonitrile, and 0.1% trifluoroacetic acid) and mobile phase B (94.9% acetonitrile, 5% ultrapure water, and 0.1% trifluoroacetic acid) were applied: 0 min, 5% B; 12 min, 45% B; 12.1 min, 100% B; 13.1 min, 100% B; and 13.2 min, 5% B. The flow rate was 0.45 ml/min over 15 min with UV detection at 214 and 498 nm.
Cell Culture
bEND3 cell line: The cells were maintained in T75 cell culture flasks at 37°C, 5% CO2, and 95% O2 (Corning, Vordingborg, Denmark) in DMEM supplemented with 10% FBS, 1 mM l-glutamine, streptomycin (90 μg/ml), and penicillin (90 U/ml) with media change every other day. The cells were sub-cultured once a week at 80% confluency using trypsin-EDTA. Cells for uptake and viability studies were seeded onto 12-well or 96-well plates (42,100 cells/cm2), respectively, and cells for live-cell microscopy were seeded onto ibidi eight-well µ-slides (ibidi GmbH, Gräfeling, Germany). The cells were ready for experimental use 48 h after seeding.
Human-induced pluripotent stem cell line BIONi010-C: The cells were maintained in Matrigel-coated six-well plates (105 cells per well) in mTeSR1 medium at 37°C, 5% CO2, and 95% O2 with media change every day. The cells were sub-cultured at 80% confluency using Versene for maintenance or Accutase for differentiation. The BIONi010-C cells were differentiated into brain capillary endothelial-like cells (Goldeman et al., 2020) using an established protocol (Stebbins et al., 2016). After 3 days of culture, the mTeSR1 medium was replaced with undifferentiated medium (UM) consisting of DMEM/F12 supplemented with 1× MEM non-essential amino acids, 1 mM GlutaMAX I, 0.1 mM β-mercaptoethanol, and 20% KOSR. The cells were cultured in UM for 6 days with daily medium changes. Thereafter, the medium was changed to hESFM supplemented with 1% PDS, bfGF (20 ng/ml), and 10 μM retinoic acid, and the cells were cultured for additional 2 days. The cells were detached from the culture plates with Accutase. Cells for uptake studies were seeded onto 12-well plates (42,100 cells/cm2). Cells for transport studies were seeded (8.9 × 105 cells/cm2) onto collagen- (400 μg/ml) and fibronectin-coated (100 μg/ml) Transwell® inserts (model 3,460, 1.12 cm2, pore size of 0.4 μm). The following day, the medium was changed to hESFM supplemented with 1% PDS and the cells were cultured for 24 h before experimental use.
In Vitro Peptide Cell Surface Adherence, Membrane Embedding, and Cellular Uptake
The bEND3 and the BIONi010-C cells grown in 12-well plates were washed twice with hHBSS at 37°C before the addition of 5 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer in hHBSS and incubation for 1 h at 37°C with orbital shaking (90 rpm). Thereafter, the cell monolayers were washed twice with 4°C hHBSS before the plates were placed on ice. The peptide fraction adhering to the plasma membrane, the peptide fraction embedded in the membrane, and the peptide fraction taken up by the cells were separated as earlier described by Frøslev et al. (Frøslev et al., 2022) using a modified protocol from Trier et al. (Trier et al., 2014). The cell surface–adhered peptide fraction was isolated by the addition of 300 µl of ice-cold 100 µM acetic acid supplemented with 15 μM sodium acetate trihydrate, 10 μM glucose, 1 μM ethylenediaminetetraacetic acid (EGTA), 1.3 μM MgSO4 heptahydrate, 5 μM KCl, 100 μM NaCl, and 1% BSA (adjusted to pH 3) for 5 min. Samples (100 µl) were taken for analysis, and the cells were washed thrice with 4°C hHBSS. The cells were transferred to −80°C for minimum 1 h. Thereafter, the cells were scraped off the bottom of the plate in the presence of 150 µl of hHBSS at 4°C and subjected to centrifugation for 20 min at 13,000 rpm and 4°C using a Sigma 1–15K centrifuge (DJB Labcare, Newport Pagnell, England) to isolate the peptide fraction taken up by the cells. Samples (100 µl) were taken from the supernatant for analysis. The remaining supernatant was discarded. hHBSS (25 µl) at 4°C was added to the cell pellet for resuspension before the addition of 75 µl of 96% ethanol to dissolve the lipid membranes. hHBSS (100 µl) at 4°C was added for volume, and the peptide fraction embedded in the membrane was isolated from the supernatant after 20 min centrifugation at 13,000 rpm and 4°C. Samples (100 µl) were taken for analysis. The samples were transferred to a black clear-bottom plate and analyzed for fluorescence using a NovoStar plate reader (BMG Labtech, Offenburg, Germany) with the gain set to 1,350 and excitation/emission wavelengths set to 485 nm/590 nm.
Peptide Uptake Into Live Cells
The cell medium was gently removed from the bEND3 cells grown in ibidi eight-well µ-slides and immediately replaced with 50 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer in hHBSS at 37°C before incubation for 1 h at 37°C with orbital shaking (90 rpm). Thereafter, the cells were gently washed twice with hHBSS at 37°C. Peptide uptake and intracellular distribution were visualized in the live cells with a Zeiss 510 laser-scanning confocal microscope (Carl Zeiss, Jena, Germany) using single-wavelength excitation at 543 nm.
Cellular Viability and Cytotoxicity
The viability of bEND3 cells grown in 96-well plates and BIONi010-C cells grown on Transwell® inserts was evaluated through assessment of the cellular metabolic activity using the sodium 3′-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis (4-methoxy-6-nitro) benzene sulfonic acid hydrate (XTT) assay according to the manufacturer’s direction. The cellular cytotoxicity of bEND3 cells grown in 96-well plates was assessed using Tryphan Blue staining.
XTT assay: The bEND3 and the BIONi010-C cells were incubated with 50 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer in hHBSS for 1 h or spiked into the cell medium for 3 h, respectively, at 37°C with orbital shaking (90 rpm). Following peptide incubation, the bEND3 and the BIONi010-C cells were gently washed twice with hHBSS or hESFM at 37°C supplemented with 1% PDS, respectively. hHBSS (bEND3) or hESFM (BIONi010-C) (100 µl) at 37°C and XTT-labeling mixture (50 µl) (labeling reagent and electron coupling reagent mixed 1:1) were added to the cells before incubation at 37°C with orbital shaking (90 rpm) for 1 h. Samples (100 µl) were transferred to a clear 96-well plate, and the absorbance was measured at 466 nm using a SPECTROstar Nano plate reader (BMG Labtech, Ortenberg, Germany). The cellular viability was calculated as follows:
where ASample is the absorbance of the test samples, ATriton X-100 is the absorbance of the dead cell control, and AhHBSS/hESFM is the absorbance of the live cell control.
Trypan Blue staining: The bEND3 cells were incubated with 50 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer in hHBSS for 1 h at 37°C with orbital shaking (90 rpm). Following peptide incubation, the cells were gently washed twice with hHBSS at 37°C before the addition of 0.2% (w/v) Trypan Blue in hHBSS at 37°C and incubation for 15 min at 37°C with orbital shaking (90 rpm). Subsequently, the cells were gently washed twice with hHBSS at 37°C, and images were obtained using a Leica DMi8 microscope (Leica Microsystems, Wetzlar, Germany) in the last washing volume.
In Vitro Peptide Transport, Cell Surface Adherence, and Cellular Uptake
The BIONi010-C monolayers were equilibrated to room temperature for 20 min, before the transendothelial electrical resistance (TEER) was measured using a Millicell-ERS Volt-Ohm meter (Fisher Scientific, Loughborough, England) equipped with an Endohm-12 cup (World Precision Instruments, Sarasota, FL, United States). The cells were re-equilibrated at 37°C on an UNIMAX 2010 shaking table (Heidolph, Schwabach, Germany). The experiment was initiated by spiking the apical chamber with T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer (final concentrations of 100 µM) or with 3 to 5-kDa fluorescein isothiocyanate (FITC) dextran (final concentration of 1 mg/ml) before 4-h incubation with orbital shaking (90 rpm). T-NR2B9c, T-Tat, and T-Tat-NR2B9c were applied alone or in the presence of C14-mannitol (0.66 μCi/ml) and H3-propranolol (1 μCi/ml). Samples (100 µl) were withdrawn from the basolateral chamber at time points 15, 30, 60, 90, 120, 180, and 240 min, with immediate replacement of the sampling volume with fresh hESFM at 37°C. Samples containing only peptide or dextran were collected in a clear-bottom black 96-well plate. TAMRA and FITC fluorescence in the samples were measured using a NovoStar plate reader (BMG Labtech, Offenburg, Germany) with at gain set to 1,350 and excitation/emission wavelengths set to 485 nm/590 nm. Samples containing H3-propranolol and C14-mannitol were transferred to scintillation vials, mixed with 2 ml of Ultima Gold 241 TM (Perkin Elmer, Waltham, MA, United States) and analyzed using a Packard Tri-Carb 2100 TR scintillation counter (Packard, Dreieich, Germany). The apparent permeability coefficient (Papp) was calculated as follows:
where dQ/dt is the steady state flux, A is the area of the filter insert (1.12 cm2), and C0 is the donor concentration in the apical chamber at time point 0 min.
After the transport study, the basolateral and apical volumes were gently replaced with fresh hESFM at 37°C, and the monolayers were equilibrated to room temperature for 20 min before TEER measurements.
For visualization of cellular peptide and dextran uptake, the cells were subjected to microscopy. The cells were fixated in 3% PFA in hHBSS for 10 min before being permeabilized with 0.1% Triton X-100 for 5 min and blocked with 2% BSA in phosphate-buffered saline (PBS) for 30 min at room temperature. The filters were removed from the inserts, and F-actin was stained by incubation with Alexa 488–conjugated phalloidin (cat no. A12378, 1:600 in PBS with 2% BSA; Thermo Fisher Scientific, Roskilde, Denmark) for 30 min at room temperature. The cells were washed three times for 5 min with 2% BSA in PBS before mounting on object slides in a drop of PBS under coverslips and sealing with nail polish. TAMRA or FITC and Alexa 488 fluorescence were imaged with single-wavelength excitation at 543 and 488 nm, respectively, using a Zeiss LSM510 confocal laser scanning microscope (Carl Zeiss, Jena, Germany). F-actin in medium control monolayers were imaged with single-wavelength excitation at 488 nm using a Leica DMi8 microscope (Leica Microsystems, Wetzlar, Germany).
For quantification of cell surface–adhered peptide and dextran, the monolayers were transferred onto ice and 300 µl of ice-cold 100 µM acetic acid supplemented with 15 μM sodium acetate trihydrate, 10 μM glucose, 1 μM EGTA, 1.3 μM MgSO4 heptahydrate, 5 μM KCl, and 100 μM NaCl, and 1% BSA (adjusted to pH 3) was added and incubated for 5 min. Samples (100 µl) were transferred to a black clear-bottom 96-well plate for analysis, and the remaining acetic acid buffer was aspirated. For isolation of peptide and dextran taken up by the cells, the filters were cut out from the filter inserts and transferred to NoStick Hydrophobic Microtubes (In Vitro A/S, Fredensborg, Denmark). Ice-cold lysis buffer (250 μl; 10 mM Tris-HCl, 0.25 M sucrose, 1 mM EDTA, 1 mM EGTA, 2% Tergitol™, and complete® inhibitor tablet) was added before 10-min incubation on ice and homogenization using a pellet pestle (Merck, St. Louis, MO, United States). The lysates were centrifuged for 10 min at 13,000 rpm and 4°C. Supernatant (100 µl) was transferred to a black clear-bottom 96-well plate for analysis. TAMRA and FITC fluorescence in the samples were measured using a NovoStar plate reader (BMG Labtech, Offenburg, Germany) with the gain set to 1,350 and excitation/emission wavelengths set to 485 nm/590 nm. The molar peptide amounts were calculated using the standard curves for each of the peptides freshly prepared in the relevant matrices.
Real-Time Barrier Integrity Measurements
The barrier integrity of the BIONi010-C monolayers was recorded in real-time using a CellZScope (NanoAnalytics, Münster, Germany). Transwell® inserts with BIONi010-C cells were transferred to the CellZScope. hESFM (1.65 ml) was added to the basolateral chamber, and the apical volume was adjusted to a total volume of 0.76 ml of hESFM before the cells were left for 30-min equilibration to 37°C followed by 60-min incubation with orbital shaking (90 rpm) at 37°C. Thereafter, T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer was spiked into the apical chamber reaching final 100 µM test concentrations with TEER measurements every 10 min for 4 h.
Immunocytochemistry and Western Blotting
T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer was spiked into the apical chamber of Transwell® grown BIONi010-C monolayers reaching final test concentrations of 100 µM. The monolayers were left for incubation with orbital shaking (90 rpm) at 37°C for 4 h. The peptide solutions were removed, and the monolayers were gently washed thrice with hHBSS at 37°C.
Monolayers for immunocytochemistry were subsequently fixed in 3% PFA in hHBSS for 10 min followed by permeabilization with 0.1% Triton X-100 for 5 min and blocking with 2% BSA in PBS for 30 min at room temperature. The filters were removed from the inserts and incubated with primary antibodies against ZO-1 (cat no. G173000, 1:100 in PBS with 2% BSA; Thermo Fisher Scientific, Roskilde, Denmark) or claudin-5 (cat no. Ab15106, 1:100 in PBS with 2% BSA; Abcam, Cambridge, United Kingdom) overnight at 4°C. The filters were washed thrice in PBS with 2% BSA before being incubated with Alexa 488–conjugated secondary antibody (cat no. AV1008, 1:100 in PBS with 2% BSA; Thermo Fisher Scientific, Roskilde, Denmark) for 30 min at room temperature. Thereafter, the filters were washed in PBS with 2% BSA and mounted on microscope slides under cover glasses using Immnu-Mount™ mounting solution (Thermo Fisher Scientific, Roskilde, Denmark). Images were acquired with single-wavelength excitation at 488 nm using a Leica DMi8 microscope (Leica Microsystems, Wetzlar, Germany).
Cells for Western blotting were lysed by incubation with Cell Extraction Buffer (cat no. FNN0011, Thermo Fisher Scientific) for 10 min on ice followed by centrifugation for 5 min at 14,000 rpm and 4°C. The total protein concentration in the supernatants was determined with the BCA assay. Samples for sodium dodecyl sulfate–polyacrylamide gel electrophoresis were prepared by mixing supernatant with loading buffer (Laemmli Buffer (Bio-Rad, Copenhagen, Denmark) supplemented with 10% β-mercaptoethanol (3:1). The samples were heated for 5 min at 95°C. Samples containing 5 µg of protein were loaded into the lanes of a 4–15% MINI-PROTEAN® TGX™ gel for separation using a Mini-PROTEAN® Tetra Cell (Bio-Rad, Copenhagen, Denmark). Precision Plus Protein™ WesternC™ Blotting Standard (5 µl) (Bio-Rad, Copenhagen, Denmark) was loaded into a separate lane. The proteins were transferred to PVDF membranes as part of a Trans-Blot® Turbo™ Mini PVDF Transfer Pack (Bio-Rad, Copenhagen, Denmark) using a Trans-Blot® Turbo Starter System (Bio-Rad, Copenhagen, Denmark). The membranes were blocked with 5% skim milk in TBS buffer (VWR Life Science, Søborg, Denmark) supplemented with 0.1% (w/v) Triton X-100 (TBS-T) and 4% BSA for 1 h at room temperature before incubation with primary antibodies in 2% skim milk in TBS-T directed against ZO-1 (cat no. Ab59720, 1:50; Cambridge, United Kingdom), claudin-5 (cat no. Ab15106, 1:100; Cambridge, United Kingdom), or β-actin (cat no. A54441, 1:10,000) overnight at 4°C. The membranes were washed three times for 5 min with TBS-T and subsequently incubated with Precision Protein Streptactin–HRP (1:1,000; Bio-Rad, Copenhagen, Denmark) and HRP-conjugated secondary antibody [cat no. G-21234 (anti-rabbit), 1:700, or cat no. 62–6,520 (anti-mouse), 1:4,000; Thermo Fisher Scientific, Roskilde, Denmark] in TBS-T with 2% skim milk for 1 h at room temperature. The membranes were washed three times for 5 min with TBS-T before visualization of the ladder and protein bands using the ECL™ Prime Western Blotting System and a Flour ChemQ imaging station (Alpha Innotech, Santa Clara, CA, United States).
Biodistribution and Immunohistochemistry
The animal studies were conducted in strict compliance with the Danish National Council for Animal Welfare under license no. 2013-15-2934-00893.
T-NR2B9c, T-Tat, T-Tat-NR2B9c, and TAMRA-T-Tat-N-dimer were dissolved in 0.9% NaCl. A 100 µl of bolus injection (3 nmol/g body weight) was administered into the tail vein of 2-week-old Sprague–Dawley rats of either sex (N = 6–9 per peptide, N = 13 for uninjected controls). The peptides were subjected to 40 min of circulation before the rats were deeply anesthetized by subcutaneous injection of 0.05 ml/10 g body weight Hypnorm/Dormicum (fentanyl/fluanisone:midazolam:sterile water ratio of 1:1:2). The total blood volume was collected from the heart before the rats were transcardially perfused with 0.01 M PBS (pH 7.4). The blood samples were centrifuged at 5,000 rcf for 30 min to pellet the cells, and the plasma was collected in clean tubes and stored at −80°C. The brain, lungs, spleen, kidney, and liver were isolated and cut into halves. Tissues for quantitative biodistribution analysis were kept at −80°C until processing. Tissues for immunohistochemical analysis (N = 2 per peptide) were fixated in 4% PFA in 0.1 M PBS (pH 7.4) overnight. Thereafter, the tissues were transferred to 30% sucrose for minnimum 48 h, before they were cryosectioned into 40-µm sections using a microtome. The tissue slices were kept in cryoprotectant solution until analysis.
Quantitative biodistribution: The tissues and blood samples were homogenized in 1 ml of ice-cold lysis buffer using a 7-ml WHEATON® Dounce Tissue Grinder (Merck, St. Louis, MO, United States) and left for 10 min on ice before being centrifuged for 10 min at 14,000 rpm and 4°C. Supernatants (100 µl) were transferred to 96-well black clear-bottom plates, and the TAMRA fluorescence in the samples was measured using a NovoStar plate reader with gain set to 1,350 and excitation/emission wavelengths set to 485 nm/590 nm. The molar peptide amounts were calculated using standard curves for each of the peptides freshly prepared in the relevant matrices.
Immunohistochemistry: All tissue sections were washed thrice with potassium-containing PBS (KPBS). Subsequently, the brain sections were blocked and permeabilized in KPBS supplemented with 3% swine serum and 0.3% Triton X-100 (blocking buffer) for 30 min at room temperature. The brain sections were thereafter incubated with primary antibody directed against CD31 (cat no. AF3628, 1:1,000; Novus Biologicals, Oxon, United Kingdom) or GFAP (cat no. ZO334, 1:500; DAKO I/S, Frederiksberg, Denmark) in blocking buffer overnight at 4°C, washed three times for 5 min in blocking buffer diluted 1:50 in KPBS, and incubated with secondary anti-goat 1:200 (cat no. A11055; Thermo Fisher Scientific, Roskilde, Denmark) or anti-rabbit antibody 1:200 (cat no. A11034; Thermo Fisher Scientific, Roskilde, Denmark) for 1 h at room temperature before washing two times for 5 min in KPBS. The brain sections as well as the lung, spleen, kidney, and liver sections were incubated with diamino-phenylindole (DAPI) (1:500) in KPBS for 5 min at room temperature and thereafter washed for 5 min in KPBS. The tissue sections were mounted on microscope slides under cover glasses using Dako Omnis fluorescence mounting media (DAKO, Glostrup, Denmark) and subjected to fluorescence microscopy using an Observer Z1 microscope with ApoTome 2 under a Plan-Apochromat ×40/1.3 Oil DIC objective or a Plan-Apochromat ×20/0.8 DIC objective (Carl Zeiss, Germany). Acquired images were corrected for brightness and contrast with ImageJ (Schindelin et al., 2012).
Data and Statistical Analysis
Microsoft Office Excell v2016 (Microsoft, Redmont, WA, United States) was used for data processing and GraphPad Prism v8 (GraphPad, La Jolla, CA, United States) for data presentation and statistical analyses. ImageJ v1.50b (Schindelin et al., 2012) was applied for Western blot analysis, and figures were prepared using Microsoft Office Power Point v2016 or Inkscape v1.1 (Inkscape, Chino Hills, CA, United States).
Data are presented as mean ± SD with N and n representing the number of technical and biological replicates, respectively. Unpaired t-test or one-way ANOVA with Tukey’s or Dunnett’s multiple comparisons test were applied. Data with p-values < 0.5 were considered statistically significant.
Results
Peptide Self-Association and/or Plasma Protein Adsorption and the Chemical Stability Differs Among the Peptide Constructs
The peptides included in the present study are listed in Figure 1A with primary amino acid sequences, molecular weights, and net charges at pH 7.4. Tat, Tat-NR2B9c, and Tat-N-dimer were N-terminally conjugated to the fluorophore TAMRA (T) to allow visualization and quantification. The chemical structures of TAMRA and Tat-N-dimer are depicted below the table (Figure 1A). Tat and other CPPs have previously been demonstrated to self-associate into soluble aggregates (Macchi et al., 2017; Kristensen et al., 2020a; Diedrichsen et al., 2021). In addition, the highly cationic nature of Tat may promote adsorption to plasma proteins, which are overall negatively charged at physiological pH (Rügheimer et al., 2008). We, therefore, investigated whether T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer self-associate and/or adsorb to plasma proteins (Figure 1B). The peptide constructs were incubated in buffer supplemented with 0.05% BSA (to hinder adsorption to the plastic ware) or in cell medium supplemented with 1% bovine serum. The solutions were subjected to centrifugal filtration through a 30-kDa cutoff membrane, which retains peptides self-associating into larger soluble aggregates and/or adsorb to plasma proteins (e.g., albumin Mw ∼69 g/mol; UniProtKB, P02769). In buffer containing low amounts of BSA, only 3.8 ± 0.1% T-NR2B9c was retained by the filter, whereas for T-Tat, T-Tat-NR2B8c, and T-Tat-N-dimer, the retention was 14.6 ± 0.1%, 35.6 ± 0.9%, and 46.4 ± 0.7%, respectively. In cell medium with 1% bovine serum, 15.4 ± 0.5% of T-NR2B9c was retained be the filter, whereas 92.6 ± 7.3%, 82.2 ± 15.5%, and 98.8 ± 2.1% of T-Tat, T-Tat-NR2B8c, and T-Tat-N-dimer, respectively, were retained. Next, the stability of the peptide constructs was evaluated during incubation in cell medium with 1% bovine serum (Figure 1C) or rat plasma (Figure 1D). T-NR2B9c and T-Tat-N-dimer displayed only little degradation in serum supplemented cell media with 88.7 ± 11% and 88.2 ± 12.8% remaining stable, respectively, after 3-h incubation. T-Tat and T-Tat-NR2B9c appeared to be more prone to degradation with 70 ± 9.5% and 50.6 ± 5.1% remaining, respectively, after 3-h incubation. In rat plasma, all peptide constructs, except T-NR2B9c, displayed substantial degradation, with only 2.0 ± 1.8% T-Tat-N-dimer remaining. T-Tat or T-Tat-NR2B9c was not detectable after 3-h incubation in rat plasma. T-Tat-NR2B9c was the least stable of the three Tat-conjugated peptides with a significantly shorter half-life (17.8 min ±3.1) than T-Tat (51.7 ±20.4 min, p < 0.05) and T-Tat-N-dimer (56.9 ±7.6 min, p < 0.05).
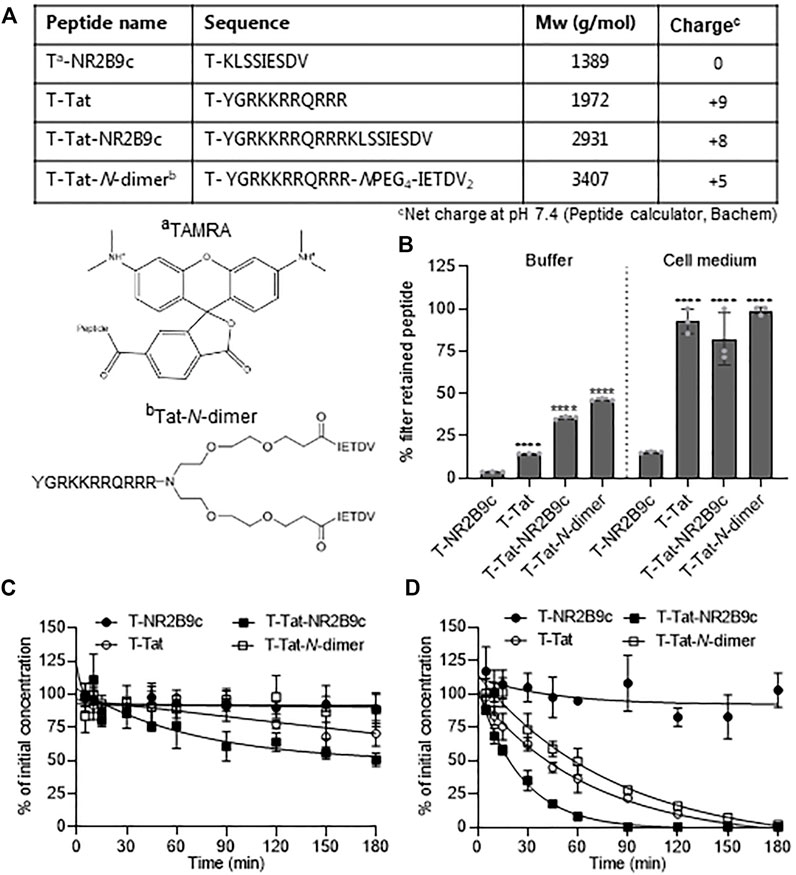
FIGURE 1. Peptide overview, stability, and aggregation/adsorption characteristics. (A) Table with primary peptide sequences, molecular weights, and net charges at pH 7.4. Chemical structure of the fluorophore TAMRA (T) and Tat-N-dimer (Bach et al., 2012). (B) Peptide self-association and/or adsorption to serum proteins assessed by centrifugal centrifugation. Mean ± SD (N = 3); ****: p < 0.0001 when compared to T-NR2B9c (one-way ANOVA with Tukey’s multiple comparisons test). Peptide stability during 3-h incubation in 37°C cell media (C) or rat plasma (D). Mean ± SD (N = 3).
The Tat-Conjugated Peptides Display Differences in Degrees of Interaction With and Uptake Into Brain Capillary Endothelial Cells but Were all Taken up by Endocytosis
The ability of T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer to interact with the endothelial plasma membrane and internalize into the brain capillary endothelial cells was studied using the mouse brain capillary endothelial cell line bEND3 (Figure 2A) and the human-induced pluripotent stem cell line BIONi010-C differentiated into brain capillary endothelial-like cells (Stebbins et al., 2016; Goldeman et al., 2020) (Figure 2B). Similar patterns in peptide cell surface adherence, cell membrane embedding, and cellular uptake were observed in the BIONi010-C–derived cells and the bEND-3 cells. T-Tat and T-Tat-NR2B9c adhered to the cell membrane to a greater extent than T-NR2B9c (p < 0.05) and T-Tat-N-dimer (p < 0.05), and a tendency toward better cell membrane embedding of T-Tat and T-Tat-NR2B9c when compared to T-NR2B9c and T-Tat-N-dimer. In contrast, all the Tat-conjugated peptides appeared to be taken up by the cells to a larger extent when compared to T-NR2B9c. Peptide uptake was studied in bEND3 cells by live cell confocal microscopy, revealing no visible uptake of T-NR2B9c (Figure 2C). On the other hand, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer all appeared to be taken up in vesicular-like structures, thus pointing toward an endocytic mechanism being responsible for their cellular uptake. Moreover, inhibition of energy-dependent uptake (i.e., endocytosis) by applying sialic acid together with T-Tat or T-Tat-NR2B9c, decreased the cellular uptake of these peptides (Supplementary Figure S1), thus supporting that endocytosis is involved in the cellular uptake of the Tat-conjugated peptides. The bEND3 cell viability, in terms of mitochondrial activity, was evaluated using the commercially available XTT assay upon incubation with T-NR2B9c, T-Tat, T-Tat-NR2B9c, T-Tat-N-dimer, or unconjugated TAMRA (Figure 2D). None of the peptides decreased bEND3 viability when compared to buffer incubated cells, whereas TAMRA alone decreased the cellular metabolic activity to just below 50%. We also studied whether T-NR2B9c, T-Tat, T-Tat-NR2B9c, T-Tat-N-dimer, or TAMRA compromised the cell membranes using Trypan Blue staining (Figure 2E). Trypan Blue dye is only permeable to cells with damaged plasma membranes. Incubation of the bEND3 cells with T-NR2B9c, T-Tat, T-Tat-N-dimer, or TAMRA did not result in cell staining, whereas Trypan Blue uptake was observed in cells incubated with T-Tat-NR2B9c.
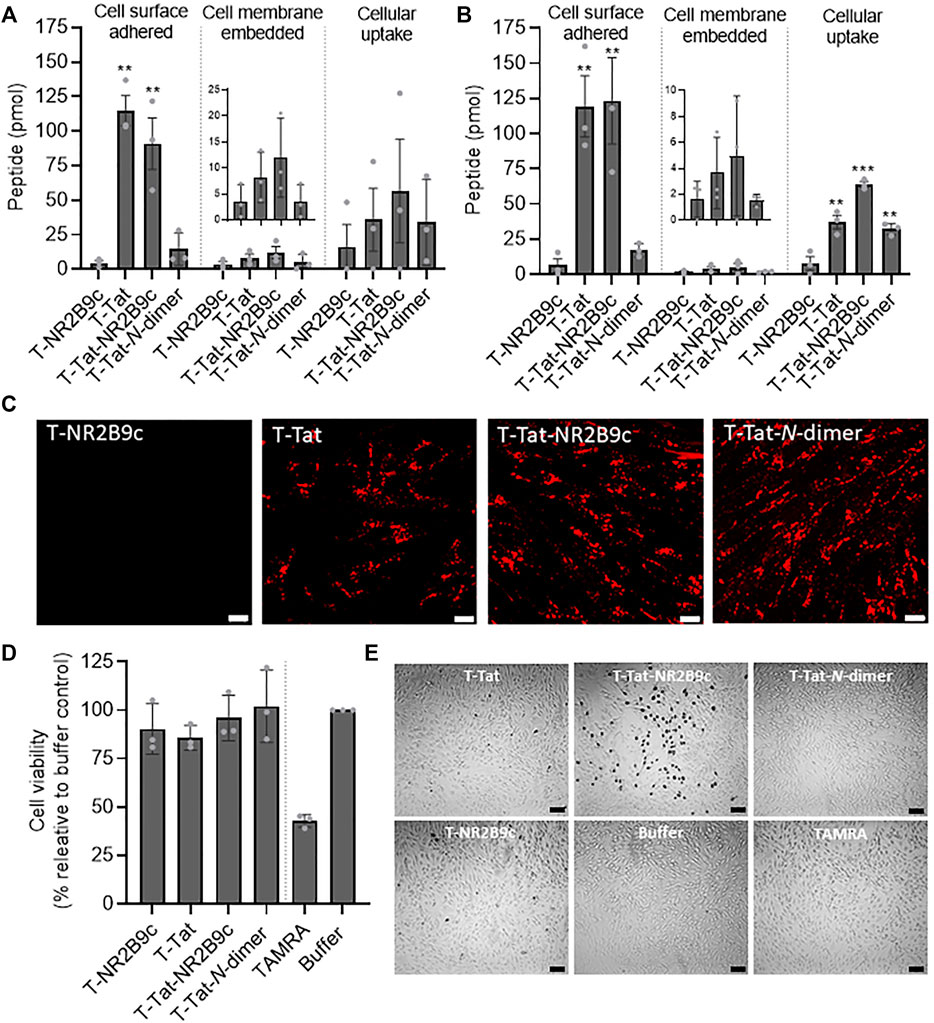
FIGURE 2. Peptide cellular distribution, live cell uptake, and effects on cellular viability. Peptide cell surface adherence, cell membrane embedding, or cellular uptake in (A) the mouse brain capillary bEND3 cell line and (B) the human-induced pluripotent stem cell line BIONi010-C after incubation with 5 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer for 1 h at 37°C. Mean ± SD (N = 3, n = 3); **: p < 0.01 and ***: p < 0.001 compared to T-NR2B9c (one-way ANOVA with Dunnett’s multiple comparisons test). (C) Peptide uptake into live bEND3 cells after incubation with 50 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer for 1 h at 37°C evaluated via TAMRA fluorescence using confocal laser-scanning microscopy. Single xy sections, scale bar: 10 µm. (D) bEND3 cell viability after incubation with 50 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, T-Tat-N-dimer, or TAMRA depicted as percentage relative buffer control. Mean ± SD (N = 3, n = 3). (E) Trypan Blue uptake into bEND3 cells after incubation with 50 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, T-Tat-N-dimer, or TAMRA imaged by bright field microscopy. Scale bar: 50 µm.
The Tat-Conjugated Peptides Were all Taken up Into BIONi010-C Cell Monolayers but Differed in the Degree of Barrier Permeation and Effects on Barrier Integrity
The ability of T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer to permeate the BBB was studied using monolayers of BIONi010-C–derived endothelial-like cells seeded onto Transwell® inserts (Figure 3A). FITC-dextran (3–5 kDa) was included as a paracellular marker reflecting the size of T-Tat-NR2B9c and T-Tat-N-dimer. Immunostainings revealed confluent monolayers via F-actin staining as well as the presence of the tight junction-associated protein ZO-1 and the BBB relevant tight junction protein claudin-5. The barrier integrity was assessed through TEER measurements before application of the peptides and FITC-dextran as well as after 4-h incubation and their subsequent removal and exchange to fresh cell culture medium (Figure 3B). The BIONi010-C monolayers displayed initial TEER-values ranging from 5,104 Ω cm2 ± 1,422 Ω cm2 to 6,240 Ω cm2 ± 160 Ω cm2, thus representing a tight barrier model (Helms et al., 2016). When comparing the initial TEER values to the TEER values measured after transport and replacement with fresh cell medium, only T-Tat-NR2B9c incubation resulted in a significant drop in TEER. After the transport study, the cell monolayers were washed and fixated before inspection for uptake of FITC-dextran, T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer via FITC or TAMRA fluorescence using confocal microscopy (Figure 3C). FITC-dextran and T-NR2B9c did not display cellular uptake, whereas T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer were all taken up by the BIONi010-C–derived cells. No conclusion on the mechanism of the Tat-mediated cellular uptake should be drawn from inspection of fixated cells due to the fact that fixation changes the intracellular CPP distribution (Richard et al., 2003). The fractions containing cell surface–adhered, cellular uptake, and transported peptide (Figure 3A) or dextran were quantified via TAMRA or FITC fluorescence, respectively (Figure 3D). Limited FITC-dextran was detected in all fractions, whereas the distribution patterns of T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer were in agreement with our earlier observations in the bEND3 cells (Figure 2A) and in the BIONi010-C–derived cells (Figure 2B) grown in the bottom of plastic wells. T-Tat and T-Tat-NR2B9c appeared superior in terms of cell surface adherence and cellular uptake when compared to T-NR2B9c and T-Tat-N-dimer. T-NR2B9c and T-Tat permeated the barrier to a higher extent than FITC-dextran. In addition, T-Tat displayed significantly better transport potential than T-NR2B9c (p < 0.01) and T-Tat-N-dimer (p < 0.05). The metabolic activity of the BIONi010-C cells was evaluated using the XTT assay and presented as cell viability in percentage relative to medium control cells (Figure 3E). None of the peptides gave rise to a decrease in cellular viability when compared to cells incubated without peptides.
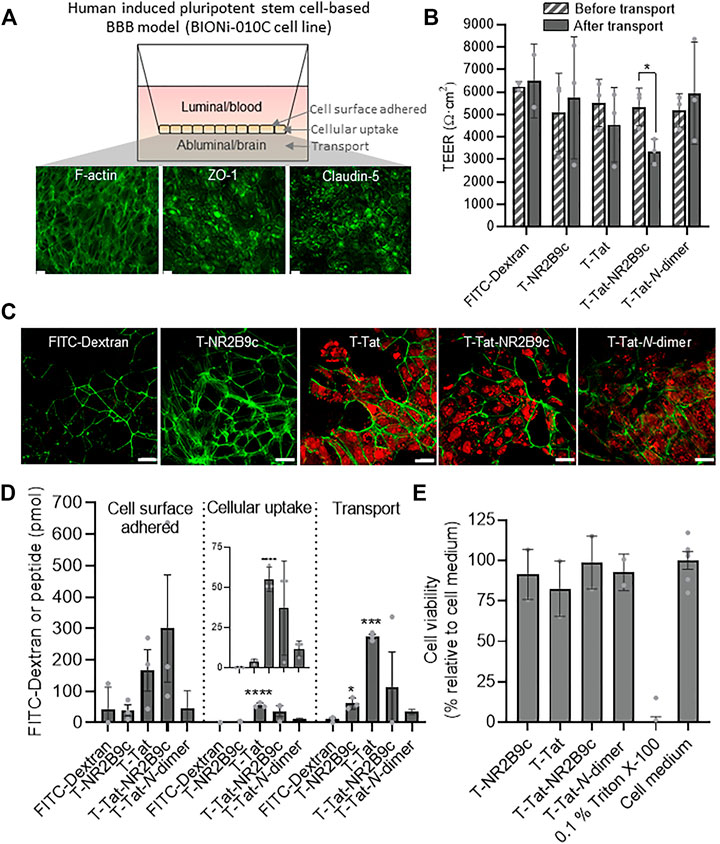
FIGURE 3. Peptide transport across and uptake into monolayers of the human pluripotent stem cell line BIONi010-C after 4-h incubation with 50 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer at 37°C. FITC-Dextran (3–5 kDa) was included as a reference paracellular marker. (A) Illustration of BIONi010-C cells differentiated into monolayers of brain capillary endothelial-like cells with representative F-actin phalloidin staining or zonula occludens-1 (ZO-1) and claudin-5 immunostainings imaged by fluorescence microscopy. Scale bar: 50 µm. (B) Transendothelial electrical resistance (TEER) measurements across BIONi010-C–derived cell monolayers before and after transport experiment. Mean ± SD (N = 3, n = 3); *: p < 0.05 (unpaired t-test) (C) Confocal laser scanning images of fixated BIONi010-C monolayers after a transport study. Dextran or peptide uptake was visualized via FITC or TAMRA fluorescence (red), respectively, whereas the cell morphology was outlined via F-actin staining using phalloidin (green). (D) Dextran and peptide cell surface adherence to, cellular uptake into, and transport across BIONi010-C monolayers. Mean ± SD (N = 3, n = 3); *: p < 0.05, ***: p < 0.001, and ****: p < 0.0001 compared to FITC-Dextran (one-way ANOVA with Tukey’s multiple comparisons test). (E) BIONi010-C cell viability depicted as percentage relative buffer control. Triton X-100 was included as control for 100% dead cells. Mean ± SD (N = 3, n = 3).
T-Tat and T-Tat-NR2B9c Decrease the Transendothelial Electrical Resistance of BIONi010-C Cell Monolayers but do not Increase Permeation of the Small Hydrophilic Marker C14-Mannitol
T-Tat-NR2B9c and, to a minor extent, T-Tat, although not significant, affected the BIONi010-C cell monolayer integrity after 4-h incubation when assessed using chamber electrodes (Figure 3B). This setup, however, does not reveal details on the barrier integrity kinetics during incubation with the peptides. We, therefore, transferred BIONi010-C cell monolayers to a CellZScope device, allowing continuous TEER measurements during peptide incubation (Figure 4A). Our transport experimental protocol includes horizontal rotation to hinder the presence of unstirred water layers as an additional barrier to peptide transport across the BIONi010-C cell monolayers. We observed a slight drop in TEER upon initiation of horizontal rotation, with the medium-incubated control cells almost reaching their initial TEER after 5.5 h. The addition of T-NR2B9c or T-Tat-N-dimer to the BIONi010-C monolayers resulted in TEER drops to 53 ± 4% and 55 ± 1% of the initial values, respectively, which for both peptides stabilized after approximately 1-h incubation. The addition of T-Tat and T-Tat-NR2B9c resulted in TEER drops to 57 ± 6% and 53 ± 4% of the initial values, which, for both peptides and in contrast to T-NR2B9c and T-Tat-N-dimer, continued to drop during incubation. The TEER of the BIONi010-C monolayers was measured in parallel using chamber electrodes before and after 4-h incubation with T-Tat, T-Tat-NR2B9c, or cell media (Figure 4B). Similar percentage drops in TEER were obtained using the CellZScope and the chamber electrodes. We investigated whether the observed drops in the measured TEER values resulting from incubation with T-Tat and T-Tat-NR2B9c affected the barrier integrity toward transport of the small hydrophilic marker C14-mannitol (182 g/mol) (Figure 4C). No enhanced C14-mannitol permeation across the BIONi010-C monolayers was observed in the presence of T-Tat or T-Tat-NR2B9c. Last, we investigated whether T-Tat and T-Tat-NR2B9c (Figure 2C) enhanced permeation of the small lipophilic marker H3-propranolol (259 g/mol) across the BIONi010-C monolayers (Figure 4D). This was not the case.
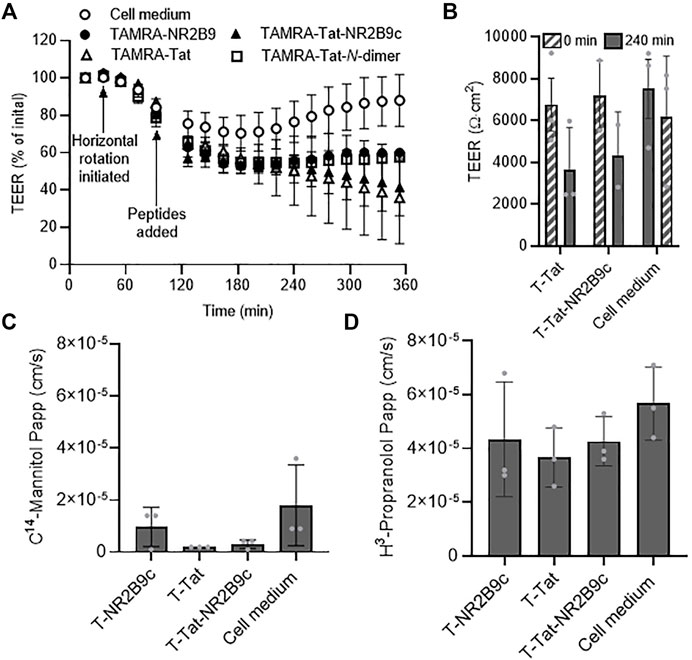
FIGURE 4. Kinetics of barrier integrity and transport of the small transcellular marker H3-propranolol or the small paracellular marker C14-mannitol during peptide incubation. (A) Real-time transendothelial electrical resistance (TEER) measurements across BIONi010-C monolayers during incubation with 50 μM T-NR2B9c, T-Tat, T-Tat-NR2B9c, or T-Tat-N-dimer for 6 h at 37°C using a CellZScope device. Mean ± SD (N = 3). (B) TEER measurements across BIONi010-C monolayers before and after incubation with 50 μM T-Tat or T-Tat-NR2B9c for 4 h at 37°C using chamber electrodes. Mean ± SD (N = 3, n = 3). (C) C14-mannitol permeability and (D) H3-propranolol permeability across BIONi010-C monolayers after 4-h incubation alone in cell media or with 50 μM T-NR2B9c, T-Tat, or T-Tat-NR2B9c at 37°C. Mean ± SD (N = 3, n = 3).
T-NR2B9c, T-Tat, and T-Tat-NR2B9c do not Affect the BIONi010-C Cell Monolayer Morphology but May Affect Tight Junction Protein Expression
We next evaluated potential effects on cell morphology and tight junction protein expression due to the observed drops in the TEER of the BIONi010-C cell monolayers after 4-h incubation with T-Tat or T-Tat-NR2B9c. F-actin-, ZO-1-, and claudin-5 protein expression patterns and levels were evaluated using immunocytochemistry (Figure 5A) and Western blotting (Figures 5B,C), respectively. F-actin staining of the BIONi010-C cell monolayers after 4-h incubation with T-NR2B9c, T-Tat, or T-Tat-NR2B9c revealed confluent monolayers (Figure 5A) with a cell morphology not differing from control monolayers incubated with cell culture media (Figure 3A). In addition, immunostainings of ZO-1 and claudin-5 indicated that the junctional alignment did not differ from the control cell monolayers. Western blot analysis was performed on cell lysates to evaluate ZO-1 and claudin-5 expression resulting from incubation with T-NR2B8c, T-Tat, or T-Tat-NR2B9c relative to their expression in cell culture medium control cells. β-Actin was included as a loading control, and bands corresponding to ZO-1 and claudin-5 were detected (Figure 5B). Evaluation of ZO-1 and claudin-5 expression levels resulting from peptide incubation relative to medium control cells did not reveal any statistical differences in protein expression among the cell lysates (Figure 5C). We did, however, observe a tendency toward lower ZO-1 expression resulting from incubation with not only T-Tat and T-tat-NR2B9c but also T-NR2B9c. In addition, the claudin-5 expression level appeared to be upregulated upon T-NR2B9c incubation when compared to the medium control cells, albeit non-significant.
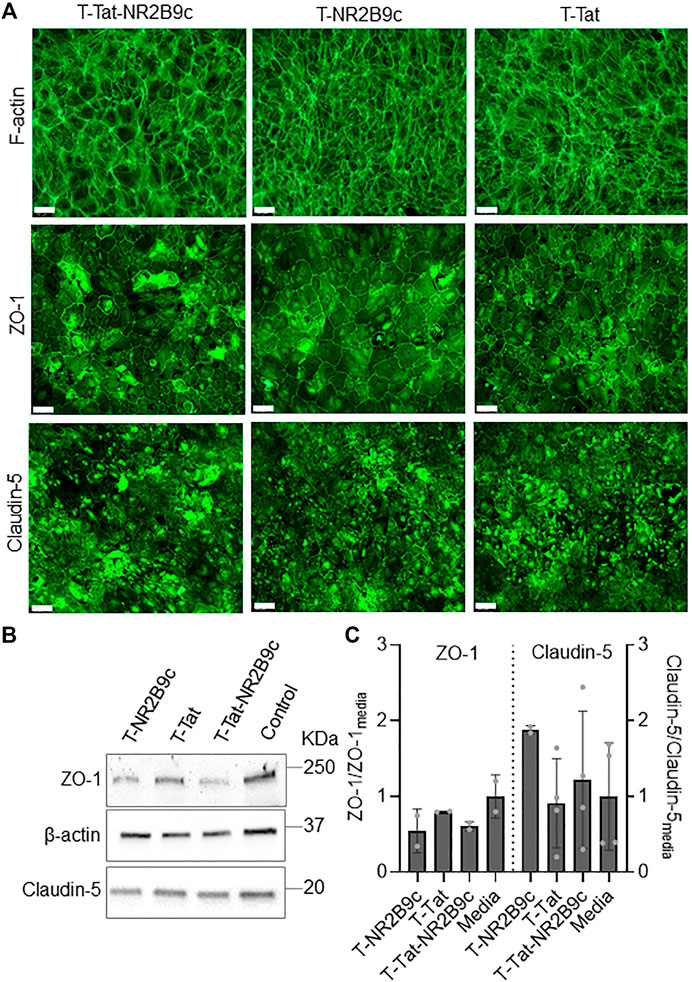
FIGURE 5. Effects on actin, zonula occludens-1 (ZO-1) and claudin-5 expression in BIONi010-C monolayers after 4-h incubation with T-NR2B9c, T-Tat, or T-Tat-NR2B9c at 37°C. (A) F-actin phalloidin staining or zonula occludens-1 (ZO-1) and claudin-5 immunostainings imaged by fluorescence microscopy. Scale bar: 50 µm. (B) Detection of ZO-1, β-actin, and claudin-5 using Western blotting. (C) ZO-1 and claudin-5 expression were quantified relative to β-actin before ZO-1 and claudin-5 band intensities from the cells incubated with T-NR2B9c, T-Tat, or T-NA-1 were standardized against the medium control cells. Mean ± SD [N = 2 (ZO-1), N = 2–4 (claudin-5)].
All Tat-Conjugated Peptides Display Broad Biodistribution in Rats but Limited Brain Accumulation
CPPs distribute widely in the organism upon systemic administration (Sarko et al., 2010; Kristensen et al., 2020b), but their potential to deliver a conjugated cargo across the BBB in vivo needs further attention. We, therefore, investigated the biodistribution of T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer after systemic administration and 40-min circulation in rats. The animals were perfused before fixation and cryosectioning. Peptide distribution was evaluated in sections of the kidney, liver, spleen, lung, and brain via the TAMRA label using fluorescence microscopy (Figure 6A). Fluorescence from the TAMRA channel was not observed in the liver, spleen, lung, and brain from blank control animals but, to a minor extent, in the kidney. In contrast, extensive TAMRA fluorescence was detected in sections of the kidney, liver, spleen, and lung from rats subjected to systemic administration of T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer. In addition, T-NR2B9c internalized in brain endothelial cells ubiquitously in the rat brain, whereas no uptake into the capillaries was observed for T-Tat. Some fluorescence was observed in brain sections from rats administered with T-Tat-NR2B9c and T-Tat-N-dimer, which, however, appeared as peptide uptake into red blood cells remaining in the capillaries due to incomplete removal during perfusion. Of note, T-NR2B9c also labeled endothelial cells of capillaries of the other organs examined beside from the brain. Brain sections were also subjected to immunostainings with the endothelial marker CD31 and the astrocyte marker GFAP to investigate potential uptake of T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer into brain capillaries and/or astrocytes (Figure 6B). Control tissue sections revealed clear staining of the endothelium as well as the astrocytes. T-NR2B9c displayed some co-localization with the endothelial marker but not with the astrocyte marker. No TAMRA fluorescence was observed in brain sections from rats subjected to T-Tat administration, whereas TAMRA fluorescence originating from T-Tat-NR2B9c and T-Tat-N-dimer again appeared as uptake into red blood cells (Figure 6B).
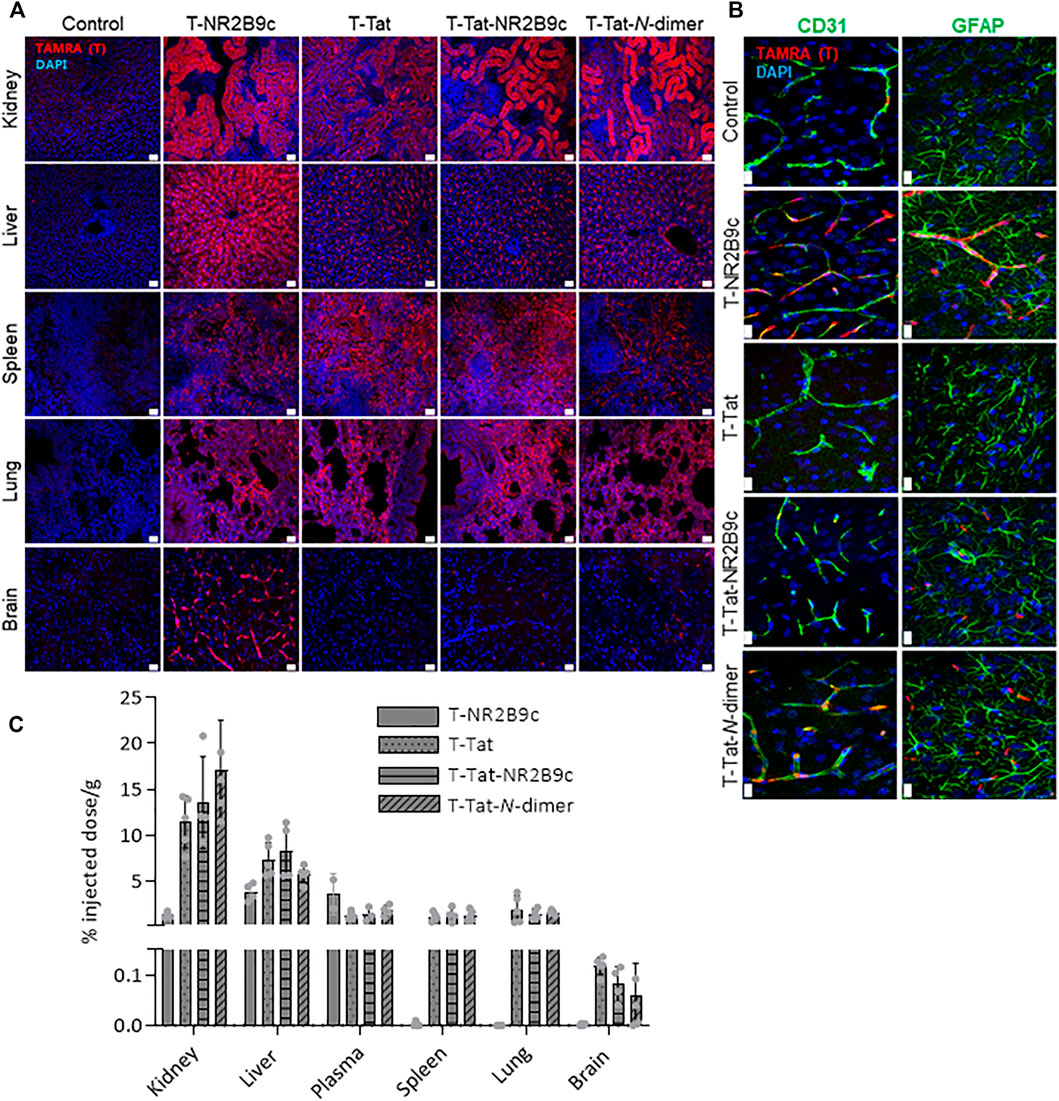
FIGURE 6. Fluorescent microscopy images of biodistribution of T-NR2B9c, T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer in rat tissue cryosections after tail vein administration of 3 nmol peptide/g body weight and 40-min circulation. (A) Peptide uptake in the kidney, liver, spleen, lung, and brain was visualized via TAMRA fluorescence (red), whereas cell nuclei were stained with DAPI (blue). (B) Brain sections were immunostained with the endothelial marker CD31 (left panel, green) and the astrocyte marker GFAP (right panel, green). Red, TAMRA; blue, DAPI. Scale bar: 20 µm. (C) Peptide uptake in the kidney, liver, spleen, lung, and brain was quantified using fluorescence spectroscopy via TAMRA fluorescence. Data are presented as % injected dose/g with mean ± SD (N = 4–5).
Last, we quantified the peptides in homogenates of the kidney, liver, plasma, spleen, lung, and brain using fluorescence spectroscopy. T-NR2B9c was detected in the kidney, liver, and plasma, but, in contrast to the immunohistochemical analysis, no T-NR2B9c was detected in the spleen and lung, whereas very limited amounts were detected in brain homogenates (0.002 ± 0.002% injected dose/g). T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer were detected in the kidney, liver, plasma, spleen, and lung, as well as, to a minor extent, in brain homogenates (0.118 ± 0.017, 0.081 ± 0.034, and 0.058 ± 0.065% injected dose/g, respectively (Figure 6C).
Discussion
Drugs that hinder ischemic stroke-induced brain tissue death are needed. The peptides Tat-NR2B9c and Tat-N-dimer are potential candidates, which have been demonstrated to reduce the infarct volume and improve cognition in mice subjected to experimental stroke (Aarts et al., 2002; Bach et al., 2012). In that sense, Tat-N-dimer displays a 1,000-fold improved target affinity and better chemical stability when compared to Tat-NR2B9c (Bach et al., 2012). However, the improved in vitro target affinity of Tat-N-dimer did not result in a similar magnitude of difference in potency upon pharmacodynamic evaluation of Tat-N-dimer and Tat-NR2B9c in mice (Bach et al., 2012) or rats (Bach et al., 2019) subjected to experimental stroke, although Tat-N-dimer is still more efficient in vivo than Tat-NR2B9c.
We recently demonstrated that T-Tat-N-dimer interacts with and permeates the BBB in vitro, to a lesser extent, than T-Tat-NR2B9c (Kristensen et al., 2020b). On the other hand, we demonstrated similar BBB permeation of the two peptides in vivo and better chemical stability of T-Tat-N-dimer in serum-supplemented cell culture media when compared to T-Tat-NR2B9c, thus questioning whether intact T-Tat-NR2B9c permeates the BBB. With this study, we aim to further understand the underlying mechanisms resulting in the observed differences in BBB permeation of Tat-NR2B9c and Tat-N-dimer.
In agreement with earlier studies (Bach et al., 2012; Kristensen et al., 2020b), we demonstrate better chemical stability of T-Tat-N-dimer in both serum-supplemented cell media (Figure 1C) and rat plasma (Figure 1D) when compared to T-Tat-NR2B9c. We moreover show that conjugation of NR2B9c or N-dimer to T-Tat lowers the ability of the resulting construct to translocate across the BIONi010-C monolayers when compared to T-Tat as a single entity (Figure 3D). Yet, T-Tat-NR2B9c and T-Tat-N-dimer internalizes into bEND3 and BIONi010-C cells more or less to similar extents as T-Tat (Figures 2A–C). The BBB endothelial cells express the NMDA receptor (Neuhaus et al., 2011). Therefore, one could speculate that T-Tat-NR2B9c and T-Tat-N-dimer, but not T-Tat, are “caught” inside the endothelial cells as a result of target engagement and thus not free for transcytosis to the abluminal side of the in vitro BBB model. However, an exact brain capillary endothelial phenotype of differentiated human-induced pluripotent stem cells, like the BIONi010-C cells, is left to be demonstrated with the current available protocols (Lu et al., 2021).
Tat and other cationic CPPs have earlier been shown to self-associate into soluble aggregates (Kristensen et al., 2015; Macchi et al., 2017; Kristensen et al., 2020a; Diedrichsen et al., 2021). Therefore, differences in the degree of self-association may add to the differences observed in BBB interaction and permeation of the peptides included in the present study. We applied centrifugal filtration to assess whether the peptides self-associate in a simple physiological buffer with 0.05% BSA and cell media with 1% serum. In the simple buffer, some T-Tat and, to a greater extent, T-Tat-NR2B9c and T-Tat-N-dimer, but not T-NR2B9c, were retained by the filter, thereby indicating primarily Tat-mediated self-association due to the limited BSA content (Figure 1B). Close to 100% of the Tat conjugates were retained by the filter in the presence of 1% serum, likely due to both peptide self-association and adsorption to plasma proteins. Plasma proteins carry an overall negative charge (Rügheimer et al., 2008) and are therefore prone to electrostatic interactions with the positively charged Tat conjugates but not with T-NR2B9c being overall neutral at physiological pH (Figure 1A). Protein corona formation was recently suggested to hamper BBB transcytosis of transferrin-decorated nanoparticles (Xiao et al., 2021). Thus, both Tat-mediated self-association and corona formation may contribute to the limited brain delivery of T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer observed in rats in the present study (Figures 6A,C). The Tat conjugates did, however, display effective uptake in peripheral tissues, which is in agreement with earlier studies (Sarko et al., 2010; Kristensen et al., 2020b). In contrast, T-NR2B9c did not self-associate in/with serum, which may, in combination with better chemical stability in rat plasma (Figure 1D), explain why only this peptide was observed in brain capillary endothelial cells in brain sections (Figure 6B). On the basis of this, it was surprisingly that T-NR2B9c displayed limited brain accumulation upon quantification in brain homogenates, when compared to T-Tat, T-Tat-NR2B9c, and T-Tat-N-dimer (Figure 6C). Because T-NR2B9c also distributed to endothelial cells of the other organs examined, a more rapid vanish may explain why the concentration was so low in blood after 40 min.
T-NR2B9c displayed limited in vitro BBB permeation when compared to T-Tat (Figure 3D). The amount of Tat-conjugated peptides detected in brain homogenates (∼0.6–0.1% of injected dose/g) is in line with earlier studies ranging 0.1–0.4% of injected dose/g (Sarko et al., 2010; Stalmans et al., 2015; Kristensen et al., 2020b). Quantification in brain homogenates, however, reflects both peptide adhering to the surface of the brain vasculature and peptide internalized into the vascular endothelial cells in addition to the peptide in the brain parenchyma and thus not necessarily peptide available for neuronal uptake for target engagement. Nevertheless, earlier studies demonstrated BBB permeation of T-Tat-NR2B9c and T-Tat-N-dimer in healthy mice using two-photon microscopy (Kucharz et al., 2017; Kristensen et al., 2020b). The different outcomes of the qualitative (Figures 6A,B) and quantitative (Figure 6C) analyses highlight the need for applying orthogonal methods to draw solid conclusions. In this case, our data rely on TAMRA fluorescence, which may be affected by the specific peptide to which it is conjugated. In addition, we are aware of the fact that the ability of the peptides to interact with and permeate the BBB likely changes upon TAMRA conjugation (Birch et al., 2017; Hedegaard et al., 2018). Optimally, future studies should include label-free detection using mass spectrometry, which, however, is challenged by the low amounts of (intact) Tat-NR2B9c and Tat-N-dimer permeating the BBB. In addition, the lack of antibodies directed against the therapeutic moieties of Tat-NR2B9c and Tat-N-dimer for immunocytochemical and immunohistochemical studies leaves us with fluorophores or alternative tags to allow visualization.
Recent studies have demonstrated that some cationic CPPs modulate the integrity of endothelial (Kristensen et al., 2020b; Frøslev et al., 2022) and epithelial (Lindgren et al., 2004; Diedrichsen et al., 2021) barriers. This suggests potential involvement of the paracellular route in addition to the more generally accepted transcytosis in CPP-mediated drug delivery across the BBB (Hervé et al., 2008; Lim et al., 2015). In the present study, we monitored the BIONi010-C cell monolayer integrity in real time and investigated whether possible Tat-mediated widening of the paracellular space would result in increased permeability of the small hydrophilic compound C14-mannitol. Incubation with T-NR2B9c, T-Tat, T-Tat-N-dimer, and T-Tat-NR2B9c resulted in a similar initial drop in TEER (∼50% of the initial value), which leveled out for monolayers incubated with T-NR2B9c and T-Tat-N-dimer, but continued to decrease upon incubation with T-Tat and T-Tat-NR2B9c (Figure 4A). The T-Tat and T-Tat-NR2B9c–mediated drop in TEER did, however, not facilitate the permeation of C14-mannitol, in contrast to another study demonstrating T-Tat-mediated C14-mannitol permeation across BIONi010-C cell monolayers (Frøslev et al., 2022). The contradictory results may be explained by the fact that the present study was performed in cell culture media supplemented with serum, whereas the earlier study was conducted in a simple HBSS buffer. In the simple buffer, T-Tat gave rise to a more dramatic drop in TEER to just below 250 Ω cm2 (∼90% of the initial value), when compared to the observation in cell culture media with measured TEER values just below 3,000 Ω cm2 (Figure 4B). A plausible explanation for these contradictory results is that T-Tat and T-Tat-NR2B9c tend to self-associate and/or adsorb to plasma protein to a greater extent in the cell media than in the simpler HBSS buffer (Figure 1B). This will, in cell culture media, hinder the Tat-moiety from interacting with the plasma membrane of the endothelial cells and eventually result in less dramatic effects on the barrier resistance when compared to studies performed in a simple buffer (Frøslev et al., 2022).
We investigated whether the T-Tat and T-Tat-NR2B9c–mediated effects on the BIONi010-C monolayer integrity were associated with changes in monolayer morphology or tight junction protein expression. The monolayer morphology was not affected, when evaluated via ZO-1 or claudin-5 immunostaining (Figure 5A), whereas a tendency toward a slight decrease in the level of ZO-1, but not claudin-5, protein expression was observed upon incubation with T-Tat and T-Tat-NR2B9c (Figure 5C). An early study documented effects on claudin-5 protein expression in primary brain microvascular endothelial cells as a result of incubation with the HIV transactivator of transcription protein Tat comprising amino acids 1–72 (Andra et al., 2003). This effect was, however, observed only after 24-h incubation with Tat1–72, but not after 3-h incubation, which is similar to the 4-h period applied in the present study, and also not resulting in changes in claudin-5 expression (Figure 5C). On the other hand, the authors did observe claudin-5 redistribution as a result of the short-term incubation with Tat, which we did not in this study (Figure 5A).
Together, we have demonstrated effective Tat-mediated uptake of the therapeutic peptides NR2B9c and N-dimer into BBB endothelial cells in vitro. We also showed that conjugation of NR2B9c or N-dimer to Tat affects the chemical stability of the resulting construct as well as its tendency to self-associate and/or adsorb to plasma proteins and thus eventually its ability to interact with and permeate the BBB. T-Tat and T-Tat-NR2B9c affected the BBB integrity in vitro but not to an extent leading to enhanced barrier permeation of the small paracellular marker C14-mannitol. Despite the ability of the Tat conjugates to internalize into the BBB endothelial cells, less efficient permeation across the healthy BBB was observed in vitro and in vivo. Thus, the earlier demonstrated therapeutic effects of Tat-NR2B9c and Tat-N-dimer in stroke animal models may be obtained due to stroke-induced effects affecting the BBB integrity (Jiang et al., 2018), thereby eventually allowing peptide permeation.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
Ethical review and approval was not required for the animal study because the animal studies were conducted in strict compliance with the Danish National Council for Animal Welfare under license no. 2013-15-2934-00893.
Author Contributions
EAH, SP, AB, CR, TM, PF, EF, and BO: Methodology, data curation, writing—review and editing. KS, AB: Resources, writing—review and editing. BB: Conceptualization, funding acquisition, writing—review and editing. MK: Conceptualization, methodology, data curation, writing—original draft, writing—review and editing, project administration.
Funding
This work was carried out within the “Research Initiative on Brain Barriers and Drug Delivery” network funded by the Lundbeck Foundation (grant no. R155-2013-14113).
Conflict of Interest
KS and AB are founders and shareholders of Avilex Pharma that develops PSD-95 inhibitors to target unmet medical needs, including stroke.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Bioneer A/S is acknowledged for providing the BIONi010-C cell line and Avilex Pharma for supplying T-Tat-N-dimer. Charlotte Goldeman Andersen and Partick Frøslev are acknowledged for cell cultivation. Alice Micklewright and Simone Amalie Kjelbæk are acknowledged for carrying out one of the bEND3 cell studies. Merete Fredsgaard and Hanne Krone Nielsen and the personnel at the animal facility, Aalborg University, Denmark, are thanked for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddev.2022.854703/full#supplementary-material
References
Aarts, M., Liu, Y., Liu, L., Besshoh, S., Arundine, M., Gurd, J. W., et al. (2002). Treatment of Ischemic Brain Damage by Perturbing NMDA Receptor- PSD-95 Protein Interactions. Science 298, 846–850. doi:10.1126/science.1072873
Andra, I. E., Pu, H., Deli, A., Nath, A., and Hennig, B. (2003). HIV-1 Tat Protein Alters Tight Junction Cultured Brain Endothelial Cells. J. Neurosci. Res. 74, 255–265. doi:10.1002/jnr.10762
Bach, A., Clausen, B. H., Kristensen, L. K., Andersen, M. G., Ellman, D. G., Hansen, P. B. L., et al. (2019). Selectivity, Efficacy and Toxicity Studies of UCCB01-144, a Dimeric Neuroprotective PSD-95 Inhibitor. Neuropharmacology 150, 100–111. doi:10.1016/j.neuropharm.2019.02.035
Bach, A., Clausen, B. H., Moller, M., Vestergaard, B., Chi, C. N., Round, A., et al. (2012). A High-Affinity, Dimeric Inhibitor of PSD-95 Bivalently Interacts with PDZ1-2 and Protects against Ischemic Brain Damage. Proc. Natl. Acad. Sci. 109, 3317–3322. doi:10.1073/pnas.1113761109
Ballarin, B., and Tymianski, M. (2018). Discovery and Development of NA-1 for the Treatment of Acute Ischemic Stroke. Acta Pharmacol. Sin. 39, 661–668. doi:10.1038/aps.2018.5
Birch, D., Christensen, M. V., Staerk, D., Franzyk, H., and Nielsen, H. M. (2017). Fluorophore Labeling of a Cell-Penetrating Peptide Induces Differential Effects on its Cellular Distribution and Affects Cell Viability. Biochim. Biophys. Acta - Biomembr. 1859, 2483–2494. doi:10.1016/j.bbamem.2017.09.015
Catanese, L., Tarsia, J., and Fisher, M. (2017). Acute Ischemic Stroke Therapy Overview. Circ. Res. 120, 541–558. doi:10.1161/CIRCRESAHA.116.309278
Cleal, K., He, L., Watson, P. D., and Jones, A. T. (2013). Endocytosis, Intracellular Traffic and Fate of Cell Penetrating Peptide Based Conjugates and Nanoparticles. Curr. Pharm. Des. 19, 2878–2894. doi:10.2174/13816128113199990297
ClinicalTrials.gov (2022b). A Phase 1 Study in Healthy Volunteers to Evaluate the Safety, Tolerability and Pharmacokinetics of AVLX-144. Available at: https://clinicaltrials.gov/ct2/show/NCT04689035 (Accessed January, 2022).
ClinicalTrials.gov (2022a). Safety and Efficacy of Nerinetide (NA-1) in Subjets Undergoing Endovascular Thrombectomy for Stroke. Available at: https://clinicaltrials.gov/ct2/show/NCT02930018 (Accessed January, 2022).
Cook, D. J., Teves, L., and Tymianski, M. (2012). Treatment of Stroke with a PSD-95 Inhibitor in the Gyrencephalic Primate Brain. Nature 483, 213–217. doi:10.1038/nature10841
Copolovici, D. M., Langel, K., Eriste, E., and Langel, Ü. (2014). Cell-penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 8, 1972–1994. doi:10.1021/nn4057269
Diedrichsen, R. G., Harloff-Helleberg, S., Werner, U., Besenius, M., Leberer, E., Kristensen, M., et al. (2021). Revealing the Importance of Carrier-Cargo Association in Delivery of Insulin and Lipidated Insulin. J. Control Release 338, 8–12. doi:10.1016/j.jconrel.2021.07.030
Frøslev, P., Franzyk, H., Ozgür, B., Brodin, B., and Kristensen, M. (2022). European Journal of Pharmaceutical Sciences Highly Cationic Cell-Penetrating Peptides Affect the Barrier Integrity and Facilitates Mannitol Permeation in a Human Stem Cell-Based Blood-Brain Barrier Model. Eur. J. Pharm. Sci. 168, 106054. doi:10.1016/j.ejps.2021.106054
Goldeman, C., Andersen, M., Al-Robai, A., Buchholtz, T., Svane, N., Ozgür, B., et al. (2020). Human Induced Pluripotent Stem Cells (BIONi010-C) Generate Tight Cell Monolayers with Blood-Brain Barrier Traits and Functional Expression of Large Neutral Amino Acid Transporter 1 (SLC7A5). Eur. J. Pharm. Sci. 156, 105577. doi:10.1016/j.ejps.2020.105577
Hedegaard, S. F., Derbas, M. S., Lind, T. K., Robertnova, M., Christensen, M. V., Michaelsen, M. H., et al. (2018). Fluorophore Labeling of a Cell- Penetrating Peptide Significantly Alters the Mode and Degree of Biomembrane Interaction. Sci. Rep. 8, 1–14. doi:10.1038/s41598-018-24154-z
Helms, H. C., Abbott, N. J., Burek, M., Cecchelli, R., Couraud, P. O., Deli, M. A., et al. (2016). In Vitro models of the Blood-Brain Barrier: An Overview of Commonly Used Brain Endothelial Cell Culture Models and Guidelines for Their Use. J. Cereb. Blood Flow Metab. 36 (5), 862–890. doi:10.1177/0271678X16630991
Hervé, F., Ghinea, N., and Scherrmann, J. M. (2008). CNS Delivery via Adsorptive Transcytosis. AAPS J. 10, 455–472. doi:10.1208/s12248-008-9055-2
Hill, M. D., Goyal, M., Menon, B. K., Nogueira, R. G., McTaggart, R. A., Demchuk, A. M., et al. (2020). Efficacy and Safety of Nerinetide for the Treatment of Acute Ischaemic Stroke (ESCAPE-NA1): A Multicentre, Double-Blind, Randomised Controlled Trial. Lancet 395, 878–887. doi:10.1016/S0140-6736(20)30258-0
Jiang, X., Andjelkovic, A. V., Zhu, L., Yang, T., Bennett, M. V. L., Chen, J., et al. (2018). Blood-brain Barrier Dysfunction and Recovery after Ischemic Stroke. Prog. Neurobiol. 163–164, 144–171. doi:10.1016/j.pneurobio.2017.10.001
Kristensen, M., Diedrichsen, R. G., Vetri, V., and Foderà, V. N. H. (2020a). Increased Carrier Peptide Stability through pH Adjustment Improves Insulin and PTH(1-34) Delivery In Vitro and In Vivo rather Than by Enforced Carrier Peptide-Cargo Complexation. Pharmaceutics 12, 993. doi:10.3390/pharmaceutics12100993
Kristensen, M., Franzyk, H., Klausen, M. T., Iversen, A., Bahnsen, J. S., Skyggebjerg, R. B., et al. (2015). Penetratin-Mediated Transepithelial Insulin Permeation: Importance of Cationic Residues and pH for Complexation and Permeation. AAPS J. 17, 1200–1209. doi:10.1208/s12248-015-9747-3
Kristensen, M., Kucharz, K., Fernandes, E. F. A., Strømgaard, K., Nielsen, M. S., Helms, H. C. C., et al. (2020b). Conjugation of Therapeutic Psd-95 Inhibitors to the Cell-Penetrating Peptide Tat Affects Blood–Brain Barrier Adherence, Uptake, and Permeation. Pharmaceutics 12, 661. doi:10.3390/pharmaceutics12070661
Kucharz, K., Søndergaard Rasmussen, I., Bach, A., Strømgaard, K., and Lauritzen, M. (2017). PSD-95 Uncoupling from NMDA Receptors by Tat- N-Dimer Ameliorates Neuronal Depolarization in Cortical Spreading Depression. J. Cereb. Blood Flow Metab. 37, 1820–1828. doi:10.1177/0271678X16645595
Lim, S., Kim, W.-J., Kim, Y.-H., Lee, S., Koo, J.-H., Lee, J.-A., et al. (2015). dNP2 Is a Blood-Brain Barrier-Permeable Peptide Enabling ctCTLA-4 Protein Delivery to Ameliorate Experimental Autoimmune Encephalomyelitis. Nat. Commun. 6, 8244. doi:10.1038/ncomms9244
Lindgren, M. E., Hällbrink, M. M., Elmquist, A. M., and Langel, U. (2004). Passage of Cell-Penetrating Peptides across a Human Epithelial Cell Layer In Vitro. Biochem. J. 377, 69–76. doi:10.1042/BJ20030760
Lu, T. M., Houghton, S., Magdeldin, T., Duran, J. G. B., Minotti, A. P., Snead, A., et al. (2021). Pluripotent Stem Cell-Derived Epithelium Misidentified as Brain Microvascular Endothelium Requires ETS Factors to Acquire Vascular Fate. Proc. Natl. Acad. Sci. U S A. 118, e2016950118. doi:10.1073/pnas.2016950118
Macchi, S., Nifosì, R., Signore, G., Di Pietro, S., Boccardi, C., D’Autilia, F., et al. (2017). Self-aggregation Propensity of Tat Peptide Revealed by UV-Vis, NMR and MD Analyses. Phys. Chem. Chem. Phys. 19, 23910–23914. doi:10.1039/C7CP04320A
Neuhaus, W., Freidl, M., Szkokan, P., Berger, M., Wirth, M., Winkler, J., et al. (2011). Effects of NMDA Receptor Modulators on a Blood-Brain Barrier In Vitro Model. Brain Res. 1394, 49–61. doi:10.1016/j.brainres.2011.04.003
Nunez, D. M., Ji, Z., Sun, X., Teves, L., Garman, J. D., and Tymianski, M. (2021). Plasmin-Resistant PSD-95 Inhibitors Resolve Effect-Modifying Drug-Drug Interactions between Alteplase and Nerinetide in Acute Stroke. Sci. Transl. Med. 13 (588), eabb1498. doi:10.1126/scitranslmed.abb1498
Rügheimer, L., Hansell, P., and Wolgast, M. (2008). Determination of the Charge of the Plasma Proteins and Consequent Donnan Equilibrium across the Capillary Barriers in the Rat Microvasculature. Acta Physiol. 194, 335–339. doi:10.1111/j.1748-1716.2008.01893.x
Richard, J. P., Melikov, K., Vives, E., Ramos, C., Verbeure, B., Gaitt, M. J., et al. (2003). B. Cell-Penetrating Peptides: A Re evaluation of the Mechanism of Cellular Uptake. J. Biol. Chem. 278, 585–90. doi:10.1074/jbc.M209548200
Sarko, D., Beijer, B., Boy, R. G., Nothelfer, E., Leotta, K., Eisenhut, M., et al. (2010). The Pharmacokinetics of Cell-Penetrating Peptides. Mol. Pharm. 7, 2224–2231. doi:10.1021/mp100223d
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 9, 676–682. doi:10.1038/nmeth.2019
Schwarze, S. R., Ho, A., Vocero-Akbani, A., and Dowdy, S. F. (1999). In Vivo protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science 285, 1569–1572. doi:10.1126/science.285.5433.1569
Sommer, J. B., Bach, A., Malá, H., Strømgaard, K., Mogensen, J., and Pickering, D. S. (2016). In Vitro and In Vivo Effects of a Novel Dimeric Inhibitor of PSD-95 on Excitotoxcity and Functional Recovery after Experimental Traumatic Brain Injury. Eur. J. Neurosci. 45, 238–248. doi:10.1111/ejn.13483
Stalmans, S., Bracke, N., Wynendaele, E., Gevaert, B., Peremans, K., Burvenich, C., et al. (2015). Cell-Penetrating Peptides Selectively Cross the Blood-Brain Barrier In Vivo. PLoS One 10, 1–22. doi:10.1371/journal.pone.0139652
Stebbins, M. J., Wilson, H. K., Canfield, S. G., Qian, T., Palecek, S. P., and Shusta, E. V. (2016). Differentiation and Characterization of Human Pluripotent Stem Cell-Derived Brain Microvascular Endothelial Cells. Methods 101, 93–102. doi:10.1016/j.ymeth.2015.10.016
Trier, S., Linderoth, L., Bjerregaard, S., Andresen, T. L., and Rahbek, U. L. (2014). Acylation of Glucagon-Like Peptide-2: Interaction with Lipid Membranes and In Vitro Intestinal Permeability. PLoS One 9 (10), e109939. doi:10.1371/journal.pone.0109939
Tünnemann, G., Martin, R. M., Haupt, S., Patsch, C., Edenhofer, F., and Cardoso, M. C. (2006). Cargo-Dependent Mode of Uptake and Bioavailability of TAT-Containing Proteins and Peptides in Living Cells. FASEB J. 20, 1775–1784. doi:10.1096/fj.05-5523com
Ugalde-triviño, L., and Margarita, D. (2021). PSD-95 : An Effective Target for Stroke Therapy Using Neuroprotective Peptides. Int. J. Mol. Sci. 22 (22), 12585. doi:10.3390/ijms222212585
Vivès, E., Brodin, P., and Lebleu, B. (1997). A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 272, 16010–16017. doi:10.1074/jbc.272.25.16010
Xiao, W., Wang, Y., Zhang, H., Liu, Y., Xie, R., He, X., et al. (2021). The Protein corona Hampers the Transcytosis of Transferrin-Modified Nanoparticles through Blood-Brain Barrier and Attenuates Their Targeting Ability to Brain Tumor. Biomaterials 274, 120888. doi:10.1016/j.biomaterials.2021.120888
Keywords: blood-brain barrer, cell-penetrating peptide, brain drug delivery, ischemic stroke, tat
Citation: Al Humaidan EL, Pedersen SL, Burkhart A, Rasmussen CLM, Moos T, Fuchs P, Fernandes EFA, Ozgür B, Strømgaard K, Bach A, Brodin B and Kristensen M (2022) The Cell-Penetrating Peptide Tat Facilitates Effective Internalization of PSD-95 Inhibitors Into Blood–Brain Barrier Endothelial Cells but less Efficient Permeation Across the Blood–Brain Barrier In Vitro and In Vivo. Front. Drug. Deliv. 2:854703. doi: 10.3389/fddev.2022.854703
Received: 14 January 2022; Accepted: 14 February 2022;
Published: 06 April 2022.
Edited by:
Siti R. Yusof, Universiti Sains Malaysia (USM), MalaysiaReviewed by:
Arsalan S. Haqqani, National Research Council Canada (NRC-CNRC), CanadaDavid Dickens, University of Liverpool, United Kingdom
Copyright © 2022 Al Humaidan, Pedersen, Burkhart, Rasmussen, Moos, Fuchs, Fernandes, Ozgür, Strømgaard, Bach, Brodin and Kristensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mie Kristensen, mie.kristensen@sund.ku.dk
†These authors have contributed equally to this work