
Abstract
Purpose
To determine if common polymorphisms in the endotoxin recognition complex influence the acute phase response as determined by the development of the systemic inflammatory response syndrome (SIRS) and platelet count on admission.
Methods
This was a prospective observational cohort study. Paediatric intensive care patients (n = 913) were genotyped for common functional polymorphisms in the endotoxin recognition complex, including Toll-like receptor 4 (TLR4). We also selected potentially confounding polymorphisms in other genes of the innate immune system. SIRS was defined by age-specific consensus criteria. Platelet counts were recorded on admission.
Results
The development of SIRS was primarily determined by the nature of the insult, but carriers of TLR4 variant alleles had lower platelet counts than children with wild-type genotype [mean ± standard error of the mean (SEM) 143 ± 7 vs. 175 ± 4; p = 0.0001)—independent of other innate immune system polymorphisms. These findings were validated using a patient cohort of 1,170 adults with coronary artery disease. Carriers of TLR4 polymorphisms with a history of myocardial infarction (n = 573) had lower platelet counts than those with the wild-type genotype (217 ± 7 vs. 237 ± 2.8; p = 0.021).
Conclusions
Our results show that TLR4 variant alleles are associated with lower platelet counts across a range of ages and precipitating insults but that they do not influence the incidence of SIRS. This result may reflect redundancy and ‘robustness’ in the pathways leading to SIRS or the lack of specificity of this endpoint. Platelet count may vary with TLR4 genotype because it may be sufficiently sensitive and more linearly related to inflammation than other markers or, alternatively, there may be a direct TLR4-mediated platelet effect.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Gram-negative bacteria cell-wall component endotoxin is a potent instigator of the inflammatory response. Lipopolysaccharide binding protein (LBP) forms complexes with the endotoxin, and these complexes are detected by the endotoxin receptor complex consisting of Toll-like receptor 4 (TLR4) and the proteins CD14 and myeloid differentiation 2 (MD2). Functional TLR4 has been described on many human cell types, including monocytes/macrophages, endothelium and platelets [1–3]. Monocyte TLR4 ligation initiates a cascade of intracellular signalling events that lead to gene transcription and the ensuing production of inflammatory mediators [Electronic Supplementary Material (ESM) Fig. E1A]. Platelet endotoxin binding is less well understood, but the mechanism includes soluble CD14 and CD62P (P-Selectin) in addition to TLR4 (ESM Fig. E1B) and contributes to thrombocytopenia [4] and neutrophil bactericidal action [5]. TLR4 has also been implicated in an endotoxin-independent response to injury, ligating fibrinogen and heat shock proteins [6].
A reduced sensitivity of the innate immune system’s capacity to recognise and respond to pathogen-associated molecular patterns has been associated with an increased frequency or severity of systemic inflammation in some studies of critically ill patients [7–9].
Two common single nucleotide polymorphisms (SNPs) exist in the human TLR4 gene: D299G and T399I. Both lie in the extra-cellular domain and are thought to reduce the effectiveness of endotoxin binding and, consequently, result in a decreased sensitivity to endotoxin [10]. The functionality of these SNPs has been observed in human studies [11] and also contested [12].
To date, only a few clinical studies have obtained convincing data that the TLR4 SNPs D299G and T399I influence the course of critical illness in general [13]. The early clinical studies that did show a difference in clinical outcome were hampered by small sample sizes (n = 77 [14], n = 159 [15], n = 91 [16]) and questionable choices in statistics [16]. Recently, more specific associations have been found between TLR4 polymorphisms and an increased vulnerability to bacteraemia in critically ill patients [17] and aspergillosis in stem cell transplantation patients [18]. The observed inconsistencies may simply reflect a lack of functional relevance of these SNPs and/or deficiencies in study design; alternatively, they may indicate that any signal from altered endotoxin recognition is drowned in the noise of other elements of host variability and environmental determinants in the acute inflammatory response. Further, the limitations of systemic inflammatory response syndrome (SIRS) as an endpoint have long-been recognised in adults [19]. This concern is compounded in the paediatric age group by the variability in the age-related normal values of the components that define SIRS (heart rate, respiratory rate and white cell count). We therefore chose a second marker of severe inflammation, namely, platelet count on admission.
Platelets actively contribute to systemic inflammation [20]. In the clinical setting, platelet count has been recognised as a sensitive and near-continuous marker of severity of critical illness in both adults [21] and children [22, 23]. Furthermore, endotoxin has been shown to directly reduce platelet count [24] to which TLR4 activity may be relevant, as shown in both mice [4] and humans [5].
We have studied 913 critically ill children to determine the relationship between altered endotoxin recognition, as identified by TLR4 SNPs, and the development of SIRS as a clinically relevant endpoint related to monocyte/macrophage activation, and platelet count on admission as an alternative marker of the acute inflammatory response. Potential confounders were assessed, including major environmental factors and other relevant SNPs in the endotoxin recognition complex and innate immune response. To validate our findings, we subsequently assessed the relationship between the TLR4 polymorphisms and platelet count on admission in a cohort of 1,170 adults with atherosclerosis. This is a well-described previously genotyped cohort with a severe inflammatory insult [25].
Methods
The Great Ormond Street Hospital for Children NHS Trust/Institute of Child Health and Southampton & South West Hampshire Local Research Ethics Committees approved this study. Parental or subject informed consent was obtained at all sites according to the Declaration of Helsinki guidelines. Clinical parameters were collected while the investigators were blinded to the genotype data and vice versa.
Subjects
Between 2000 and 2006 children were recruited consecutively in three recruiting time-periods from three tertiary paediatric intensive care units. Inclusion and exclusion criteria have been documented previously [8].
Subjects were divided by primary diagnosis into ‘Bypass’, ‘Infection’ and ‘Non-infection’ groups. Briefly, children who were admitted for heart surgery on cardiopulmonary bypass were assigned to ‘Bypass’; those children who were diagnosed with an infectious process by the admitting physician were assigned ‘Infection’; the category ‘Non-infection’ was used for all other admission diagnoses, mainly trauma and elective surgery (see ESM for details).
Identification of polymorphisms
A literature search performed to identify functional common SNPs in the endotoxin recognition complex resulted in the identification of TLR4 D299G and TLR4 T399I and the CD14 promoter −159C > T. LBP P97P was included as a common SNP of unknown functionality that may influence outcome from sepsis. No common SNPs were identified in MD2.
Possible confounding SNPs in the following genes and/or promoters are listed in Table 1: mannose binding lectin (MBL-2), cholinergic receptor, nicotinic, alpha 7 (CHRNA7), interleukin (IL)-6, IL-10, tumor necrosis factor alpha (TNFα) and human plasminogen activator inhibitor-1 (PAI-1). These SNPs have either been shown to have an effect on the acute inflammatory response or are thought to have modifier effects on TLR4 activity (see ESM for details on rationale and genotyping methods).
Outcome measures
The main outcome measures were the dichotomous measure ‘development of SIRS in the first 3 days of intensive care stay’ and the near-continuous variable ‘platelet count on admission’. SIRS was defined according to the 1992 American College of Chest Physicians/Society of Critical Care Medicine (ACCP/SCCM) Consensus Conference [26], with adjustments as described previously [8, 9].
The duration of ventilation, length of stay and development of nosocomial infection in the first week of intensive care stay were secondary outcome measures.
Validation cohort
A cohort of 1,170 Caucasian patients, angiographically diagnosed to have coronary artery stenosis (>50% stenosis in ≥1 major epicardial coronary artery) in the Southampton Atherosclerosis Study, was recruited and genotyped for the TLR4 polymorphism D299G. Genotype and phenotype are described in detail elsewhere [25]. A subset of patients (n = 777) was also genotyped by Taqman analysis for both the D299G and T399I polymorphisms. Given the substantial time and financial constraints associated with recruiting a large equivalent cohort, we chose this cohort as a readily available relevant dataset.
Based on our observations in critically ill children, we hypothesised that TLR4 variant carriers with a history of recent myocardial infarction as a severity threshold for inflammation would show lower platelet counts on admission.
Statistical analysis
Hardy–Weinberg equilibrium was assessed by the χ2 test. Univariate analysis was performed using the χ2, Mann–Whitney U, Student’s t test or one-way analysis of variance (ANOVA) as appropriate. Genotype parameters were included in a binomial regression analysis in addition to clinically relevant parameters. A two-tailed p value <0.05 was considered to be statistically significant. All statistical analyses were performed using SPSS ver. 13.0 for Mac OS X (SPSS, Chicago, IL).
Results
Genotyping
In total, we successfully genotyped 906 (99.2%) critically ill children.
All SNPs were in Hardy–Weinberg equilibrium according to ethnic background, and allele frequencies were in accordance with the literature [27].
TLR4 genotype distribution differed between ethnic groups. The children of Asian descent (n = 26) exhibited only the wild-type TLR4 genotype, whereas amongst those of African descent (n = 78) the TLR4 D299G polymorphism was common (n = 10, 12%) and T399I was observed only once (1.2%). In contrast, both variant alleles were present [D299G, n = 72 (9.6%); T399I, n = 74 (9.9%)] in the Caucasian group (n = 747) and they usually occurred concurrently, i.e., showed cosegregation (ESM Fig. E2).
Development of SIRS in the first 3 days of intensive care stay
SIRS occurred in 594/913 (65%) patients. The prevalence of early SIRS varied between the different diagnostic groups, with significantly more children in the Infection group developing SIRS (143/179, 79.8%) than in the Non-infection (126/199, 63.3%) or Bypass groups (325/535, 60.7%) (Table 2).
No significant difference was observed in the development of SIRS between subjects with wild-type or variant genotypes for either of the TLR4 variant alleles alone or in combination; 66% (68/103) of the subjects with a TLR4 mutation developed SIRS versus 65% (522/803) with the wild-type genotype (Fig. 1a).

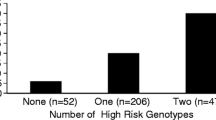
a Univariate analysis for the Toll-like receptor 4 (TLR4) polymorphism variant alleles as a risk factor for systemic inflammatory response syndrome (SIRS) in the total paediatric cohort. The p value is derived from the χ2 test. b Platelet count for the total paediatric cohort stratified by TLR4 genotype—wild-type versus variants. p value is derived from Student’s t test. c One-way analysis of variance (ANOVA) for platelet count per TLR4 variant allele (0,1 or ≥2) for the total paediatric cohort. d Thrombocytopenia (platelet count < 150 × 109) stratified by TLR4 genotype for the total paediatric cohort [odds ratio (OR) 1.7, 95% confidence interval (CI) 1.1–2.6; χ2 test)]. SEM Standard error of the mean
In the Bypass or Infection groups (ESM Figs. E3A, E4A), the incidence of SIRS did not differ when stratified according to TLR4 genotypes. In the Non-infection group, however, we noted a trend for carriers of the TLR4 variant alleles to be at an increased risk for developing SIRS [odds ratio (OR) 2.4, 95% confidence interval (CI) 0.84–6.6; p = 0.09] (ESM Fig. E5A). After multivariate analysis adjusting for basic demographics and other polymorphisms in the endotoxin recognition complex, such as the CHRNA7, PAI-1, MBL-2 and genes encoding cytokines IL-6, TNFa and IL-10 this just reached statistical significance (p = 0.03) with OR 5.4 (95% CI 1.1–26.5).
Platelet count
In the total paediatric cohort there was a marked difference in admission platelet count between patients with wild-type and those carrying the 299G and/or 399I TLR4 variant [mean ± standard error of the mean (SEM) 175 ± 4 vs. 143 ± 7 × 109/L, respectively; p = 0.0001] (Fig. 1b). This effect was additive (Fig. 1c). Consequently, the proportion of children with actual thrombocytopenia (platelet count <150 × 109/L) was significantly higher amongst those carrying one or more variant TLR4 alleles compared to those with the wild-type genotype (OR 1.7, 95% CI 1.1–2.7; p = 0.01) (Fig. 1d; Table 3).
Interestingly, the effect that TLR4 polymorphisms exerted differed between the precipitating event groups. No difference in platelet count could be seen in the Bypass group (ESM Fig. E3B), while in the Non-infection group, children with variant TLR4 alleles had lower admission platelet counts than those with the wild-type genotype (ESM Fig. E5B). A trend in the same direction was also evident in the smaller Infection group (ESM Fig. E4B).
The risk for thrombocytopenia on admission in the whole population associated with TLR4 variants remained after multiple regression analysis. It was independent of any combination of primary diagnosis, demographic characteristics, neutrophil count, SIRS, PIM score and other endotoxin complex and PAI-1, MBL-2, CHRNA7, IL-6, IL-10 or TNFα genotypes (OR 2.2, 95% CI 1.2–3.9; p = 0.01).
Polymorphisms in LBP or CD14 did not alter the risk for the development of SIRS or platelet count (ESM Table E3).
The secondary clinical outcome measures, i.e. length of stay, length of ventilation or nosocomial infection, did not show any stratification according to genotype (ESM Table E4).
Validation cohort
Both phenotypic data [age 63.3 ± 0.3 years (mean ± SEM), sex ratio (M/F) 3.2, 888/276] and TLR4 D299G genotype (in Hardy–Weinberg equilibrium) were available for 1,065 patients. Of these patients, 573 (54%) had experienced a recent (within 3 months) myocardial infarction. In this latter group, those subjects with TLR4 variant genotypes had lower platelet counts than those with the wild-type genotype (mean ± SEM: 217 ± 7.0 vs. 237 ± 2.8 × 109/L; p = 0.021; Fig. 2). Because of the high prevalence of allele cosegregation in this Caucasian cohort, this study was not powered to assess a ‘gene dose effect’. There was no significant difference in platelet count in the group that had not suffered a recent myocardial infarction (variant vs. wild-type genotype: 244 ± 10 vs. 229 ± 3.5 × 109/L; p = 0.14).

Platelet count for adults with a history of myocardial infarction, stratified by TLR4 genotype (wild-type vs. variant). p value is derived from Student’s t test
Discussion
In this cohort of more than 900 critically ill children we found little impact of common functional polymorphisms in the endotoxin receptor on the development of SIRS. Conversely, we were able to show a clear and unequivocal effect of TLR4 genotype on platelet count. Polymorphisms associated with reduced TLR4 function were found to exert a dose-dependent effect, with the lowest platelet counts found in those haplotypes associated with the lowest TLR4 function. The stepwise association between TLR4 polymorphisms and lower platelet counts may imply that these two SNPs reduce TLR4 function by addition or interaction.
We have also confirmed that common co-segregation for the D299G and T399I polymorphisms is found only in Caucasians. This result indicates that studies analysing either of these SNPs in isolation will be suboptimal in multiethnic cohorts [28]. This factor may underlie some of the variability in the results reported in published studies.
What does a clinically small (175 vs. 143 × 109/L) but statistically highly significant difference in platelet count associated with TLR4 haplotype mean given the lack of effect on SIRS? These observations appear to be contradictory, but perhaps they offer some insight into acute inflammatory processes and the limitations of the current definitions of systemic inflammation employed for clinical research.
The apparent conflict arises from the view that platelets are an integral part of the acute inflammatory response [29] and that there is an important relationship between thrombocytopenia and severity of illness across all age ranges [21, 22, 30, 31]. Numerous observations suggest that platelets actively contribute to the intensity of the acute inflammatory response rather than being bystanders that are consumed as a ‘para-phenomenon’ [32, 33]. The suggested mechanisms include an acceleration of the recruitment of neutrophils and monocytes through the formation of heterotypic complexes as well as the production of inflammatory mediators, including CD40 ligand, tissue factor, RANTES and matrix metalloproteinases [34].
If platelet activation is inseparable from the acute inflammatory response, why might reduced endotoxin responsiveness affect platelet count but have no detectable effect on clinical markers of systemic inflammation? The simplest explanation would be a type II error. SIRS may be an insufficiently precise measure and, therefore, despite the numbers of cases reported here, this study may be inadequately powered. The limitations of a clinical diagnosis of SIRS are well documented [19]. In part this is due to therapy-induced changes on the parameters temperature, heart rate and respiratory rate that constitute clinical SIRS. The label SIRS may thus poorly reflect the underlying state of immune activation. Also, we have only studied children with a precipitating insult of sufficient severity to require admission to intensive care for organ support. Any variability in the development of SIRS that may be attributable to TLR4 polymorphisms may be swamped by the severity of this level of insult.
The fact that SIRS is a binary outcome measure compounds these limitations. In contrast, platelet count on admission is a near continuous measure that is unlikely to be confounded dramatically by therapy. Alternatively, it may be that platelet count is just a more sensitive measure of the degree of inflammation than the crude indices used to define SIRS.
Importantly, no influence of MBL-2 genotype on platelet count was demonstrated, while others and our group have previously described an increased risk of SIRS in children carrying MBL-2 genotypes associated with reduced plasma levels of the pattern recognition molecule MBL. Therefore, another explanation for the discordant results between the impact of TLR4 polymorphisms on the development of SIRS and platelet count requires consideration. Rather than these results reflect the degree of complexity and specificity of the two end-points themselves, it may be that they reflect a difference in the level of complexity of the inflammatory pathways relevant to the two end-points. The development of SIRS is a complex process with multiple pathways and, therefore, a high level of redundancy. As such, the inherited dysfunction or therapeutic inhibition of single elements are unlikely to have a large impact on overall outcome [35, 36]. Specifically, the intra-cellular signalling response to TLR ligation has been described in great detail as an example of a biologically robust process [37]. In contrast to the main effector cells of systemic inflammation (monocyte/macrophages and endothelial cells), platelets express a very limited range of surface receptors for inflammatory mediators and have a limited response repertoire due to the absence of genomic DNA [33]. Thus, it is biologically plausible that endotoxin-TLR4 binding causes a direct, non-redundant response of platelet activation and consumption that is independent of the development of SIRS.
The demonstration of functional TLR4 expression on platelets [3, 38] and the observation that adults with TLR4 variant genotypes have significant differences in platelet function [39] provide some support for this concept.
We sought to validate this idea in an independent cohort with a relevant inflammatory insult: reduced severity of atherosclerosis has previously been associated with TLR4 variant genotypes [39, 40]. We were indeed able to show that adults with coronary artery stenosis and a recent history of myocardial infarction who carried TLR4 variant alleles had platelets counts which, on average, were 20 × 109/L lower than those of their wild-type counterparts.
Finally, clinical trial evidence further supports the possibility of the impact of this pathway on platelet count. In a double blind multi-centre randomised study of 393 children with severe meningococcal disease, the anti-endotoxin molecule recombinant bactericidal/permeability-increasing protein fragment (rBPI21) had no significant effects on outcome, but thrombocytopenia was less severe in patients receiving rBPI21 (control group 36% transfused platelets vs. 25% r-BPI21 group, p = 0.03) [41].
Our study has several limitations. The paediatric cohort is heterogeneous. Different precipitating events in nature and time course before admission may have led to our inability to determine a differentiation in SIRS. Given the inherent non-protocolised nature of this observational study, we did not attempt to describe SIRS by way of humoral biomarkers of inflammation, such as cytokines. Future strictly protocolised studies may elucidate the temporal pattern of circulating cytokines in relation to the genetic profile.
The validation cohort differed in age, inflammatory insult and ethnic mix and was, therefore, not a perfect match. However, myocardial infarction is an inflammatory process of substantial severity with a relevance to platelet count and TLR4. Despite this imperfect match, the small (although statistically significant) difference in platelet count and the fact that counts were in the normal range, this result still corroborates our findings in the paediatric cohort.
Polymorphisms in endogenous bactericidal/permeability-increasing protein (BPI) [42] and at least two components of TLR4-induced intracellular signalling, such as Mal [43] and IRAK4 [44], may change susceptibility or outcome in severe infections. We did not type for these polymorphisms, and thus our study might be labelled as incomplete. However, given the ever-evolving knowledge in this area, all similar studies are hampered by this phenomenon.
Last, we did not confirm our findings in human ex vivo experiments or in vivo knock-out mice models to determine the underlying processes. Future studies will need to focus on the specific underlying pathophysiological mechanisms.
In conclusion, we have shown that TLR4 polymorphisms are associated with lower platelet counts in severe inflammation. The reasons for this are unclear but may point to a direct effect of the TLR4 pathways on platelets or indicate that platelet counts are a more sensitive marker of systemic inflammation than SIRS criteria. These data support the view that variation in TLR4 function influences the early inflammatory response.
References
Zarember KA, Godowski PJ (2002) Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 168:554–561
Dauphinee SM, Karsan A (2006) Lipopolysaccharide signaling in endothelial cells. Lab Invest 86:9–22
Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P (2005) Platelets express functional Toll-like receptor-4. Blood 106:2417–2423
Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, Ni H, Lazarus AH, Freedman J, Semple JW (2006) Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 107:637–641
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13:463–469
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21:335–376
Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR, Abraham E (2006) Variant IRAK-1 haplotype is associated with increased nuclear factor-kappaB activation and worse outcomes in sepsis. Am J Respir Crit Care Med 173:1335–1341
Fidler KJ, Wilson P, Davies JC, Turner MW, Peters MJ, Klein NJ (2004) Increased incidence and severity of the systemic inflammatory response syndrome in patients deficient in mannose-binding lectin. Intensive Care Med 30:1438–1445
Stephens RC, Fidler K, Wilson P, Barclay GR, Mythen MG, Dixon GL, Turner MW, Klein NJ, Peters MJ (2006) Endotoxin immunity and the development of the systemic inflammatory response syndrome in critically ill children. Intensive Care Med 32:286–294
Rallabhandi P, Bell J, Boukhvalova MS, Medvedev A, Lorenz E, Arditi M, Hemming VG, Blanco JC, Segal DM, Vogel SN (2006) Analysis of TLR4 polymorphic variants: new insights into TLR4/MD-2/CD14 stoichiometry, structure, and signaling. J Immunol 177:322–332
Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA (2000) TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 25:187–191
van der Graaf C, Kullberg BJ, Joosten L, Verver-Jansen T, Jacobs L, van der Meer JW, Netea MG (2005) Functional consequences of the Asp299Gly Toll-like receptor-4 polymorphism. Cytokine 30:264–268
Imahara SD, O’Keefe GE (2004) Genetic determinants of the inflammatory response. Curr Opin Crit Care 10:318–324
Agnese DM, Calvano JE, Hahm SJ, Coyle SM, Corbett SA, Calvano SE, Lowry SF (2002) Human toll-like receptor 4 mutations but not CD14 polymorphisms are associated with an increased risk of Gram-negative infections. J Infect Dis 186:1522–1525
Barber RC, Aragaki CC, Rivera-Chavez FA, Purdue GF, Hunt JL, Horton JW (2004) TLR4 and TNF-alpha polymorphisms are associated with an increased risk for severe sepsis following burn injury. J Med Genet 41:808–813
Lorenz E, Mira JP, Frees KL, Schwartz DA (2002) Relevance of mutations in the TLR4 receptor in patients with gram-negative septic shock. Arch Intern Med 162:1028–1032
Henckaerts L, Nielsen KR, Steffensen R, Van Steen K, Mathieu C, Giulietti A, Wouters PJ, Milants I, Vanhorebeek I, Langouche L, Vermeire S, Rutgeerts P, Thiel S, Wilmer A, Hansen TK, Van den Berghe G (2009) Polymorphisms in innate immunity genes predispose to bacteremia and death in the medical intensive care unit. Crit Care Med 37(192–201):e191–e193
Bochud PY, Chien JW, Marr KA, Leisenring WM, Upton A, Janer M, Rodrigues SD, Li S, Hansen JA, Zhao LP, Aderem A, Boeckh M (2008) Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N Engl J Med 359:1766–1777
Vincent JL (1997) Dear SIRS, I’m sorry to say that I don’t like you. Crit Care Med 25:372–374
Levi M, Lowenberg EC (2008) Thrombocytopenia in critically ill patients. Semin Thromb Hemost 34:417–424
Akca S, Haji-Michael P, de Mendonca A, Suter P, Levi M, Vincent JL (2002) Time course of platelet counts in critically ill patients. Crit Care Med 30:753–756
Peters MJ, Ross-Russell RI, White D, Kerr SJ, Eaton FE, Keengwe IN, Tasker RC, Wade AM, Klein NJ (2001) Early severe neutropenia and thrombocytopenia identifies the highest risk cases of severe meningococcal disease. Pediatr Crit Care Med 2:225–231
Nguyen TC, Carcillo JA (2006) Bench-to-bedside review: thrombocytopenia-associated multiple organ failure—a newly appreciated syndrome in the critically ill. Crit Care 10:235
Ohtaki Y, Shimauchi H, Yokochi T, Takada H, Endo Y (2002) In vivo platelet response to lipopolysaccharide in mice: proposed method for evaluating new antiplatelet drugs. Thromb Res 108:303–309
Yang IA, Holloway JW, Ye S (2003) TLR4 Asp299Gly polymorphism is not associated with coronary artery stenosis. Atherosclerosis 170:187–190
American College of Chest Physicians/Society of Critical Care Medicine (1992) American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 20:864–874
Smirnova I, Hamblin MT, McBride C, Beutler B, Di RA (2001) Excess of rare amino acid polymorphisms in the Toll-like receptor 4 in humans. Genetics 158:1657–1664
Ferwerda B, McCall MB, Verheijen K, Kullberg BJ, van der Ven AJ, Van der Meer JW, Netea MG (2008) Functional consequences of Toll-like receptor 4 polymorphisms. Mol Med 14:346–352
Klinger MH, Jelkmann W (2002) Role of blood platelets in infection and inflammation. J Interferon Cytokine Res 22:913–922
Vanderschueren S, De Weerdt A, Malbrain M, Vankersschaever D, Frans E, Wilmer A, Bobbaers H (2000) Thrombocytopenia and prognosis in intensive care. Crit Care Med 28:1871–1876
Ragazzi S, Pierro A, Peters M, Fasoli L, Eaton S (2003) Early full blood count and severity of disease in neonates with necrotizing enterocolitis. Pediatr Surg Int 19:376–379
Zarbock A, Polanowska-Grabowska RK, Ley K (2007) Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev 21:99–111
Davi G, Patrono C (2007) Platelet activation and atherothrombosis. N Engl J Med 357:2482–2494
Gawaz M, Langer H, May AE (2005) Platelets in inflammation and atherogenesis. J Clin Invest 115:3378–3384
Zeni F, Freeman B, Natanson C (1997) Anti-inflammatory therapies to treat sepsis and septic shock: a reassessment. Crit Care Med 25:1095–1100
Bone RC (1996) Why sepsis trials fail. JAMA 276:565–566
Oda K, Kitano H (2006) A comprehensive map of the toll-like receptor signaling network. Mol Syst Biol 2:2006.0015
Shashkin PN, Brown GT, Ghosh A, Marathe GK, McIntyre TM (2008) Lipopolysaccharide is a direct agonist for platelet RNA splicing. J Immunol 181:3495–3502
Patrignani P, Di Febbo C, Tacconelli S, Moretta V, Baccante G, Sciulli MG, Ricciotti E, Capone ML, Antonucci I, Guglielmi MD, Stuppia L, Porreca E (2006) Reduced thromboxane biosynthesis in carriers of toll-like receptor 4 polymorphisms in vivo. Blood 107:3572–3574
Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA (2002) Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med 347:185–192
Levin M, Quint PA, Goldstein B, Barton P, Bradley JS, Shemie SD, Yeh T, Kim SS, Cafaro DP, Scannon PJ, Giroir BP (2000) Recombinant bactericidal/permeability-increasing protein (rBPI21) as adjunctive treatment for children with severe meningococcal sepsis: a randomised trial. rBPI21 Meningococcal Sepsis Study Group. Lancet 356:961–967
Michalek J, Svetlikova P, Fedora M, Klimovic M, Klapacova L, Bartosova D, Elbl L, Hrstkova H, Hubacek JA (2007) Bactericidal permeability increasing protein gene variants in children with sepsis. Intensive Care Med 33:2158–2164
Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, Ling EY, Frodsham AJ, Walley AJ, Kyrieleis O, Khan A, Aucan C, Segal S, Moore CE, Knox K, Campbell SJ, Lienhardt C, Scott A, Aaby P, Sow OY, Grignani RT, Sillah J, Sirugo G, Peshu N, Williams TN, Maitland K, Davies RJ, Kwiatkowski DP, Day NP, Yala D, Crook DW, Marsh K, Berkley JA, O’Neill LA, Hill AV (2007) A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet 39:523–528
Ku CL, von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, Chrabieh M, Issekutz AC, Cunningham CK, Gallin J, Holland SM, Roifman C, Ehl S, Smart J, Tang M, Barrat FJ, Levy O, McDonald D, Day-Good NK, Miller R, Takada H, Hara T, Al-Hajjar S, Al-Ghonaium A, Speert D, Sanlaville D, Li X, Geissmann F, Vivier E, Marodi L, Garty BZ, Chapel H, Rodriguez-Gallego C, Bossuyt X, Abel L, Puel A, Casanova JL (2007) Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med 204:2407–2422
Baldini M, Lohman IC, Halonen M, Erickson RP, Holt PG, Martinez FD (1999) A Polymorphism* in the 5′ flanking region of the CD14 gene is associated with circulating soluble CD14 levels and with total serum immunoglobulin E. Am J Respir Cell Mol Biol 20:976–983
Hubacek JA, Skodova Z, Adamkova V, Lanska V, Vlasakova Z, Poledne R (2004) Association of the −159C –> T polymorphism in the CD14 promoter with variations in serum lipoproteins in healthy subjects. Blood Coagul Fibrinolysis 15:365–366
Hubacek JA, Stuber F, Frohlich D, Book M, Wetegrove S, Ritter M, Rothe G, Schmitz G (2001) Gene variants of the bactericidal/permeability increasing protein and lipopolysaccharide binding protein in sepsis patients: gender-specific genetic predisposition to sepsis. Crit Care Med 29:557–561
Barber RC, O’Keefe GE (2003) Characterization of a single nucleotide polymorphism in the lipopolysaccharide binding protein and its association with sepsis. Am J Respir Crit Care Med 167:1316–1320
Shann F, Pearson G, Slater A, Wilkinson K (1997) Paediatric index of mortality (PIM): a mortality prediction model for children in intensive care. Intensive Care Med 23:201–207
Acknowledgements
We thank Helen Tighe, Annette Mcquillan, Kerry Illing, Deepan Sivakumar and Leanne Clifford for data collection, Matthew Rose-Zerilli for genotyping, Hugh Montgomery and Aroon Hingoraani for expert advice and patients and parents for their willingness to enrol their children in the study. This work was undertaken at Great Ormond Street Hospital/UCL Institute of Child Health, which received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. Dr R.S. Agbeko was supported by the Special Trustees Great Ormond Street Hospital. Dr K. Fidler was funded by a Wellcome Clinical Training Fellowship. Dr J. Holloway received support from the Southampton Medical and Dental Federation and the Asthma, Allergy and Inflammation Research Charity. Dr J. Deanfield and Dr S. Ye acknowledge support by The British Heart Foundation.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Agbeko, R.S., Holloway, J.W., Allen, M.L. et al. Genetic polymorphisms in the endotoxin receptor may influence platelet count as part of the acute phase response in critically ill children. Intensive Care Med 36, 1023–1032 (2010). https://doi.org/10.1007/s00134-010-1857-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-010-1857-x