
Abstract
Haploinsufficiency of TRIP12 causes a neurodevelopmental disorder characterized by intellectual disability associated with epilepsy, autism spectrum disorder and dysmorphic features, also named Clark-Baraitser syndrome. Only a limited number of cases have been reported to date. We aimed to further delineate the TRIP12-associated phenotype and objectify characteristic facial traits through GestaltMatcher image analysis based on deep-learning algorithms in order to establish a TRIP12 gestalt. 38 individuals between 3 and 66 years (F = 20, M = 18) - 1 previously published and 37 novel individuals - were recruited through an ERN ITHACA call for collaboration. 35 TRIP12 variants were identified, including frameshift (n = 15) and nonsense (n = 6) variants, as well as missense (n = 5) and splice (n = 3) variants, intragenic deletions (n = 4) and two multigene deletions disrupting TRIP12. Though variable in severity, global developmental delay was noted in all individuals, with language deficit most pronounced. About half showed autistic features and susceptibility to obesity seemed inherent to this disorder. A more severe expression was noted in individuals with a missense variant. Facial analysis showed a clear gestalt including deep-set eyes with narrow palpebral fissures and fullness of the upper eyelids, downturned corners of the mouth and large, often low-set ears with prominent earlobes. We report the largest cohort to date of individuals with TRIP12 variants, further delineating the associated phenotype and introducing a facial gestalt. These findings will improve future counseling and patient guidance.
Similar content being viewed by others


Introduction
TRIP12-associated intellectual disability was initially described by Clark and Baraitser in 1987 as an X-linked intellectual disability syndrome in a mother and her two sons with intellectual disability (ID), obesity and dysmorphic features [1]. Genome and targeted sequencing on archived DNA revealed a heterozygous likely pathogenic nonsense variant NM_004238.3:c.2983 C > T in the TRIP12 gene at chromosomal location 2q36.3 in all three subjects [2], consistent with an autosomal dominant inheritance. In the last years, TRIP12 was postulated as a candidate gene for autism spectrum disorder (ASD) and ID in large, multigene or exome-based studies [3,4,5], but most of these lack detailed clinical descriptions.
Bramswig et al. published the first cohort of TRIP12 individuals, providing detailed information on 11 individuals harboring loss-of-function TRIP12 variants [6]. Recurrent clinical features included mild to moderate ID, ASD, behavioral anomalies and craniofacial dysmorphism without a distinct facial phenotype. Seizures were present in a minority of individuals. No clear genotype-phenotype correlations were observed and haploinsufficiency was proposed as the underlying pathogenic mechanism. Zhang et al. expanded the clinical and mutational spectrum by reporting a further 9 unrelated individuals with pathogenic inactivating TRIP12 variants, of which 5 carried a copy number variant (CNV) affecting TRIP12 [7]. All individuals presented with a childhood-onset neurodevelopmental disorder associated with ID varying from moderate to severe with language delay and verbal deficiency more pronounced than previously reported. Obesity was present in one third of these subjects and those reported by Bramswig et al. combined [6, 7]. A similar phenotype was reported in two additional male individuals by Donoghue et al., as well as in individuals carrying larger intragenic variants or deletions encompassing TRIP12 at the 2q36.3 locus [8].
TRIP12 (Thyroid hormone Receptor-Interacting Protein 12 (OMIM *604506)) encodes a ubiquitin ligase and is part of the E3 ubiquitin ligase family. This group contains over 500 different proteins implicated in various rare and more common neurodegenerative and neurodevelopmental disorders [9]. TRIP12 contains four different non-unique protein domains: a catalytic domain homologous to the E6-AP C-Terminus (HECT), two protein/protein interaction domains (WWE (Tryptophan-Tryptophan-Glutamate) and ARM (Armadillo repeats)), and an Intrinsically Disordered Region (IDR). Through binding with different protein interactors it has a role in various molecular pathways such as cell cycle progression, DNA damage response, chromatin remodeling and cell differentiation [10].
Here, we further establish the neurodevelopmental phenotype of TRIP12 haploinsufficiency by reporting the hitherto largest cohort of TRIP12 individuals, including 1 previously published and 37 novel individuals, carrying 34 novel and 1 previously reported variants. In addition, we describe a TRIP12-gestalt through facial analysis of 21 individuals using GestaltMatcher [11].
Materials and methods
Patients and recruitment
Clinical and genotypic information on 38 different individuals from 37 unrelated families was obtained. Individuals were recruited through a European Reference Network ITHACA call for collaboration and include individuals from Belgium, Denmark, Estonia, France, Italy, Luxembourg, The Netherlands, Germany, Spain and Switzerland. Appropriate informed consent was obtained for affected individuals, in accordance with the local ethics committee. Subject 26 was previously published at age 7 years [7], but updated phenotypic information at age 10 was collected.
Genotypic and clinical information was collected through an information table and included developmental and neurological features, medical history, physical signs and brain magnetic resonance imaging (MRI) data, when available. Written informed consent for the publication of pictures was obtained for 21 individuals. These were uploaded into the GestaltMatcher Database (GMDB) for phenotypic analysis of facial morphology [11].
Computational facial analysis
GestaltMatcher [11] is an extension of DeepGestalt and quantifies the facial syndromic similarity between two patients. First, GestaltMatcher was trained on 3438 frontal pictures of patients with 139 different disorders from the GestaltMatcher Database to learn syndromic facial features. It then encoded each of the 21 TRIP12 pictures into a 320-dimensional facial phenotype descriptor. These facial phenotype descriptors formed a 320-dimensional Clinical Face Phenotype Space (CFPS). A cosine distance was calculated in the CFPS to objectify the facial syndromic similarity between two pictures. A small distance between two pictures correlates to a high facial syndromic similarity. The pictures were ranked in ascending order based on the cosine distance. To investigate the facial similarity of TRIP12 subjects compared to other disorders, 4278 individuals with 257 different disorders were selected and assigned to the GestaltMatcher gallery to simulate a real-world scenario. Then, a pairwise comparison was performed by iterating the following steps for each TRIP12 picture: [1] selection of one TRIP12 test picture; [2] assignment of the other 20 TRIP12 pictures to the GestaltMatcher gallery; [3] calculation of the ranks of the other 20 pictures. Findings were visualized in a pairwise rank matrix.
Next, an average healthy and TRIP12 facial image were established. For every TRIP12 facial picture (n = 21), facial pictures of 10 healthy UTKFace [12] individuals were selected (n = 210). These were matched for gender and age of the TRIP12 individual. An average healthy and TRIP12 face was then generated based on alignment of facial landmarks, followed by averaging across all pictures.
Molecular investigations
Most TRIP12 variants were identified by massive parallel sequencing techniques, such as gene panel sequencing (n = 10), exome sequencing (n = 20) and genome sequencing (n = 1). Family segregation of the paternally inherited variant in subject 2 was confirmed by Sanger sequencing. Chromosomal microarray analysis was used for the detection of the larger deletions. No other relevant candidate variants were detected by these techniques in any of the individuals. All TRIP12 variants were uploaded to the DECIPHER database (DECIPHER Patient ID 489845-489882) [13].
Results
Variants
Variant information
For an overview of detailed clinical and molecular information, we refer to Supplementary Table 1. We report on 35 (34 novel and 1 previously described) variants affecting TRIP12 (visualized in Fig. 1). All are described according to the transcript NM_004238.3, that encodes isoform c of the protein (NP_004229.1).

These include (A) 34 novel (top) and 20 previously reported [6,7,8] (bottom) TRIP12 single-nucleotide variants or indels. Colors represent different variant types: missense (green), frameshift (blue), nonsense (red), small in-frame deletion (purple), splice (orange), translocation (grey) and (B) 5 novel (red) and 3 previously reported [7] (black) larger deletions affecting TRIP12.
Variants were distributed throughout the entire gene and its corresponding protein domains. There were no clear variant hotspots though variants in the WWE domain were overrepresented when corrected for domain length (ratio of variants/domain length: 0.034 compared to 0.018, 0.014 and 0.013 in the ARM, HECT and IDR domain respectively).
Haploinsufficiency has been proposed as the underlying pathogenic mechanism for the TRIP12-associated neurodevelopmental disorder [6]. Of the 35 reported variants here, 15 concerned a frameshift variant (43%), predicted to lead to the incorporation of a premature stop codon. These were found throughout the entire protein. One frameshift variant NM_004238.3:c.273dup was detected in two independent families (subjects 9 and 10).
Nonsense variants (n = 6) accounted for 17% of described TRIP12 pathogenic variants and were mainly detected in the proximal half of the protein. One nonsense variant NM_004238.3:c.1507 C > T was identified in two different individuals (subjects 4 and 5). Thus far, no pathogenic nonsense variants have been reported in the HECT domain.
All 3 splice variants (subjects 25, 26 and 27) consisted of a G to A substitution at the +1 position of the donor splice site of exons 38, 25 and 19 respectively.
One of these, NM_004238.3:c.3743 + 1 G > A (subject 26), was previously reported (Zhang et al., subject 9) and quantitative PCR showed a reduction of TRIP12 mRNA levels in the proband compared to the unaffected family members, assuming that this splice variant results in nonsense-mediated decay [7].
For the other two splice variants, no functional research was performed, though three independent in silico prediction tools (SSF [14], MaxEnt [15] and NNSPLICE [16]) showed loss of the donor splice site.
Intragenic TRIP12 deletions were found in 4 different individuals (11% of variants). Of these, 3 were multi-exon deletions ranging from 5.9 kb to 26.1 kb. The other was a small in-frame deletion of 9 nucleotides in exon 10; NM_004238.3:c.1503_1511del (subject 33). In the absence of functional studies, the latter variant was initially classified as a variant of uncertain significance (class 3) according to the ACMG guidelines [17]. However, DNA methylation episignature analysis showed clustering of this sample with TRIP12 cases and allowed reclassification of this variant as (likely) pathogenic [18]. No intragenic deletions identified here or previously, were located within the region encoding the WWE protein domain.
Previously reported intragenic TRIP12 deletions are visualized in Fig. 1B [7]. Larger deletions encompassing TRIP12 are not included except for two novel deletions in subjects 34 and 38.
Both deletions lead to TRIP12 disruption, hereby presumably abolishing normal protein function. The deletion in subject 34 contains two other genes, but these are not associated with and OMIM morbid phenotype and have a low probability of loss of function intolerance (pLI). Besides TRIP12, the deletion in subject 38 affects 34 genes, of which 21 are protein coding, but of all genes linked to an OMIM morbid phenotype in this region, only TRIP12 is predicted to be loss-of-function intolerant (pLI = 1).
Finally, 5 novel de novo missense variants (14%) were identified. No functional studies have been conducted to assess the effect of these variants on protein function, but DNA methylation analysis provided evidence for pathogenicity of the missense variant in subject 32 and identified subject 29 as an outlier, segregating with controls instead of other TRIP12 individuals [18]. No DNA methylation studies were performed for the variants in subjects 28, 30 and 31, and so these were classified as variants of uncertain significance. All previously reported (n = 5) and newly reported missense variants are located within a protein domain (IDR, WWE or HECT domain) with the exception of the NM_004238.3:c.3676 C > T variant in subject 30 (interdomain region between WWE and HECT domain).
Inheritance
De novo occurrence was confirmed in most cases where parental testing was performed (31/33). Subject 7 was mosaic for the NM_004238.3:c.2458 C > T nonsense variant, but did not seem to present a milder phenotype. The variant was found in 28/219 reads by gene panel analysis, corresponding to a mosaicism level of approximately 26% and later confirmed in 38% by Sanger sequencing. In 2 individuals a pathogenic variant was inherited from an affected parent. The NM_004238.3:c.1132 C > T nonsense variant arose de novo in the father (subject 1) and was subsequently inherited by his son (subject 2). This is the only reported case in which a TRIP12 variant was inherited from an affected father. There was no information on possible mosaicism in subject 1.
In subject 10 the NM_004238.3:c.273dup frameshift variant was inherited from his mildly affected mother (not included in this cohort). She presented with speech delay, but no additional information was provided. The same variant was also detected in subject 9. Here, the variant was absent in the subject’s healthy mother, but no testing of the father was performed.
Familial occurrence of a pathogenic TRIP12 variant generally involves less severe phenotypes. Indeed, subjects 1, 2 and 10 showed only mild intellectual disability in the absence of associated behavioral problems, autism or seizures. In addition, the facial features were rather subtle in these cases.
These findings are consistent with two previous cases of familial occurrence. Mild intellectual disability was reported in the affected mother of two more severely impaired sons [1]. Bramswig et al. described a boy (individual 6) with mild developmental delay (attended regular school until the age of 12 years), a typical autism and ADHD [6]. He inherited a missense variant from his healthy mother, corresponding to either reduced penetrance of this disorder, mosaicism in the mother or questionable pathogenicity of the missense variant.
Summary of clinical findings
The main clinical characteristics of the 38 individuals reported here (Fig. 2 and Supplementary Table 1) include variable developmental delay and intellectual disability marked by important speech delay, autism, obesity and distinct facial features. Supplementary Table 1 also summarizes the clinical findings in previously reported individuals with intragenic TRIP12 variants. The two individuals harboring a deletion encompassing TRIP12, as well as several adjacent genes reported by Zhang et al. were not included, nor Subjects 8 and 9 as they were reported by Bramswig et al. and updated in this cohort respectively [6, 7].

Grey: total cohort (n = 38), light blue: proportion of assessed individuals, dark blue: proportion of affected individuals.
Prenatal period and early childhood
Pregnancy was uneventful in more than 80% of cases, resulting in a term birth in about 80%. Two pregnancies were complicated by intrauterine growth retardation (IUGR) with preterm delivery. In one, IUGR was accompanied by oligohydramnios (subject 19), leading to severe postnatal growth restriction at age 7 (weight −3.2 SD, height −2.3 SD and head circumference −3 SD). In the other, only mild IUGR was noted (subject 16) and biometry at age 6 was within the low normal range (weight −1.9 SD, height −1.8 SD and head circumference −1.8 SD).
Recurrent infections occurred in about 30% of subjects during early childhood and mainly involved ear- and respiratory infections.
Neurodevelopmental phenotype
Motor development was marked by an important delay in more than 85% of subjects, but remained within the mild range in most. All except for one, managed unassisted walking before the age of 4. Subject 28 was more severely affected. She never achieved independent walking and was wheelchair-bound at age 6 years. Persistent motor problems into childhood were noticed in 70% and primarily concerned fine motor skills. Childhood hypotonia was present in 53%, but represents a rather nonspecific finding. In some individuals (subjects 22, 28 and 33) progression of neurological symptoms into spasticity with muscle hypertonia and pyramidal reflexes was observed.
Intellectual disability was present in 100%, and proved to be highly variable, ranging from mild to severe (IQ range 40–79, mean IQ 58). More than half of the subjects had moderate intellectual abilities, consistent with earlier reporting.
All individuals presented speech delay, one of the hallmarks of the TRIP12 neurodevelopmental phenotype. Rarely, individuals acquired relatively high-level verbal skills with normal speech (splice variant in subject 26) and the ability to read and write (missense variant of uncertain significance segregating with episignature controls in subject 29 [18]), but language skills remained limited in most. This contrasts with those reported by Bramswig et al. of whom 6/11 spoke fluently. Here, speech in the form of short and simple sentences was achieved by 19/33 individuals, but 6 individuals only obtained single word language and 6 remained completely nonverbal, communicating through sign language or not at all.
The TRIP12-intellectual disability phenotype has been previously linked to variable behavioral difficulties. In this cohort 76% of individuals showed some form of behavioral problems ranging from insecurity and impaired social skills to rebellious behavior with aggression and self-harm. Sleeping problems (21%) were characterized by frequent nocturnal awakenings. Autistic features, present in half of the cohort, were less prevalent than previously reported (76%).
Six individuals had movement disorders, of whom most showed stereotypies. Other more rare neurological findings included ataxia, gait abnormalities (unsteady, toe walk) and spasticity.
Five individuals (13%) experienced epileptic seizures (subjects 4, 6, 28, 30 and 35). This is consistent with earlier findings, where about 20% experienced childhood-onset seizures. Epilepsy was most severe in subjects 28 and 30, both carrying a missense variant. In subject 28, infantile spasms started at 7 months of age, later evolving to generalized seizures with hypsarrhythmia on EEG. Despite treatment with multiple antiepileptic drugs and the need for a vagal nerve stimulator, the epilepsy in this girl remained refractory at the age of 15 years. Starting at 6 years of age, subject 30 presented with recurrent tonic-clonic seizures, correlating to parietal lobe seizures on EEG. Anti-epileptic treatment was not further specified, but epilepsy in this subject remained refractory at age 44. The other individuals showed a somewhat milder epileptic presentation.
A period of regression was noted in about 10% of individuals (subjects 9, 23, 29, 31 and 33) and occurred either in infancy (at 6 months in subject 31 and at 16 months following hospital admission in subject 9) or at a later age (at 63 years in subjects 23 and 29). However, in subject 29 cognitive decline due to a possible stroke could not be excluded. Subject 33 spoke her first sentences at age 4, followed by a language regression one year later. Occurrence of regression has been described previously [6, 8].
Biometry
Growth parameters were largely within normal range, but overweight was a recurrent feature in almost half of the subjects. When corrected for age, body-mass indexes in these subjects were within the range of overweight in 8/16, of obesity in 7/16 and of extreme obesity in one subject [19]. Excess weight seemed due to hyperphagia or other eating disorders in multiple cases. Consistent with previous reports, weight gain began in childhood as early as 2 years of age.
Phenotype in elderly
Most previously reported TRIP12 individuals are children, with the oldest being 26 years old. In our study, 11 subjects were older than 18 years and 3 individuals were 50 + (subject 6 was 52 years, subject 23 was 65 years and subject 29 was 66 years). Hormonal imbalances were present in the two eldest individuals with hypothyroidism, osteoporosis and frequent fractures, but no other important health problems were noticed. Although both individuals experienced cognitive decline at age 63 years, it is not clear whether this was due to a subclinical cerebrovascular incident or part of a premature aging process inherent to the TRIP12-associated disorder.
Miscellaneous
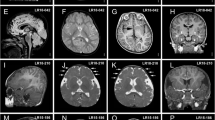
Magnetic resonance brain imaging showed nonspecific abnormalities in about half of the individuals. This is not completely in line with previous findings, where 15/16 brain MRIs were reported as normal (Supplementary Table 1).
Subtle hand and foot abnormalities were noted in 30% and 24% of the subjects, respectively and mainly included brachy- and clinodactyly of the fifth digit. No important internal anomalies were observed in this cohort nor have been reported elsewhere.
Facial gestalt
The facial features (Fig. 3) of TRIP12 individuals are variable, though, contrary to previously thought, often recognizable. Deep-set eyes, noted in 70% of individuals in this cohort and 100% of individuals reported previously, form one of the hallmark facial characteristics of this disorder. Other recurrent eye features included fullness of the upper eyelids (45%), narrow palpebral fissures (44%) and epicanthus (38%).

From left to right and top to bottom; subject 1 at 16 years, subject 2 at 1 year 7 months, subject 4 at 23 years 6 months, subject 10 at 5 years 8 months, subject 11 at 2 years 11 months, subject 12 at 18 years 5 months, subject 13 at 8 years 5 months, subject 14 at 5 years 9 months, subject 17 at 7 years, subject 18 at 4 years 3 months, subject 21 at 9 years, subject 23 at 65 years, subject 24 at 10 years, subject 26 at 7 years, subject 27 at 22 years, subject 28 at 14 years 8 months, subject 31 at 4 years 2 months, subject 34 at 19 years, subject 35 at 13 years 6 months, subject 37 at 2 years 10 months, subject 38 at 4 years 9 months. Recurrent facial features include deep-set eyes with narrow palpebral fissures, fullness of the upper eyelids, a broad nasal tip and wide mouth with prominent Cupid’s bow and downturned corners. Ears are often large and low-set with prominent earlobes.
The nose was characterized by a broad nasal tip (70%) with low columella (50%) and depressed nasal bridge (36%).
The mouth was more variable and often wide (44%) with downturned corners in several individuals. A Cupid’s bow (52%), thin upper lip vermillion (42%) and long philtrum (32%) were significantly more prevalent here than previously reported (24%, 18% and 12%, respectively).
Ears were earlier described as small and posteriorly rotated, but seemed rather large in our cohort. Consistent with previous findings, low ear implantation was seen in 32%, accompanied by large earlobes in 42%.
GestaltMatcher analysis
Characteristic facial features as described above were confirmed in the averaging facial analysis, showing a distinct TRIP12 gestalt compared to healthy individuals (Fig. 4).

A Healthy average face based on 210–10 for each TRIP12 individual (n = 21) – UTKFace individuals matched for gender and age and (B) TRIP12 average face showing characteristic facial features, marked by deep-set eyes, full upper eyelids, a broad nasal tip and a wide mouth with prominent Cupid’s bow.
The pairwise ranks of the 21 TRIP12 pictures (Fig. 5) also showed a similar facial phenotype among most of the individuals compared to the 4,278 individuals with 257 different disorders from the GestaltMatcher Database. 6/21 pictures had at least one TRIP12 individual in their top-10 rank, and 14/21 had at least one in their top-30 rank. Two phenotypic clusters (column of subject 1 to 38 and column of subject 27 to 34) were distinguished. Subject 23 was an outlier, but this might be due to his advanced age. Although subjects 1 and 2 were father and son respectively, their facial features were not very similar (ranks 363 and 381). This might also be due to the difference in age, which is a known confounder of GestaltMatcher. No clear clusters based on the type of variant were observed, possibly due to the limited number of facial pictures available for GestaltMatcher analysis (Fig. 5).

Every column/row represents one TRIP12 subject (labeled with their subject number and corresponding variant type). TRIP12 pictures were matched in the GestaltMatcher gallery (4278 plus 20 TRIP12 pictures) and each TRIP12 picture was ranked based on the cosine distance, calculated in the CFPS. For example, when testing subject 34, subject 35 was at the 13th rank and subject 12 at the 24th rank. The red boxes highlight the two clusters of subjects with the highest facial similarities, but no correlation with the variant type is observed.
Genotype-phenotype correlations
Localization
As illustrated in Fig. 1, TRIP12 variants are distributed throughout the entire gene and its corresponding protein domains, as well as in the interdomain regions. Though slightly more frequent in the WWE domain when corrected for length, there are no clear mutation hotspots. Moreover, phenotypic severity does not seem to correlate with variant localization.
Variant type
Subjects 9 and 10 carried the same TRIP12 frameshift variant NM_004238.3:c.273dup. Similarly, subjects 4 and 5 both harbored the NM_004238.3:c.1507 C > T nonsense variant in the ARM region of TRIP12 and finally subjects 1 and 2 were father and son, carrying the NM_004238.3:c.1132 C > T nonsense variant. Even though the phenotype is largely similar in the different subjects harboring the same TRIP12 variant, some inter- and intrafamilial variability can be seen pointing towards other genetic as well as environmental contributing factors.
Missense variants seem to be associated with a more severe phenotypic presentation, although the total cohort is still small (n = 10). Regression was more frequent and a similar trend was observed for ASD and epilepsy, the latter more refractory in individuals with a missense variant. Intellectual and language levels were generally lower and the only individual who never achieved independent walking (subject 28) carried a missense variant. Remarkably, obesity was seen in 23/53 individuals (43%) but was less frequent in individuals with a missense variant (3/10, 30%).
All missense variants but one were located in a functional TRIP12 protein domain, and although functional evidence is lacking, we could speculate that these variants might result in an alternative pathomechanism explaining the more severe phenotype. However, TRIP12 DNA methylation analysis of subject 29 by van der Laan et al. (Case 21) showed clustering of this sample with controls, indicating questionable causality of this variant [18].
A small intragenic in-frame deletion of 9 nucleotides (subject 33) was classified as (likely) pathogenic after DNA methylation episignature analysis [18]. Given the more severe clinical phenotype (Supplementary Table 1), this deletion might also exert a missense-like effect, though this remains to be further elucidated functionally.
As expected, facial dysmorphism was more subtle in all three individuals harboring a splice variant (no consent for publication was obtained for subject 25), who also exhibited a better cognitive development. It is conceivable that in these individuals some protein function is retained, hence leading to a less pronounced presentation.
In addition to intragenic variants, we also report two larger deletions disrupting TRIP12 at intron 7 and exon 34 (subjects 34 and 38), deleting the proximal and distal exons respectively, as well as several adjacent genes. The two additional genes, FBXO36 and SLC16A14, involved in the deletion in subject 34 were not linked to an OMIM phenotype and TRIP12 was the only gene with a high pLI score (pLI = 1 [0−1]), indicating extreme intolerance to loss of function. Based on the depletion of loss-of-function variants in gnomAD (observed <10% of expected), this score confirmed the haploinsufficient character of TRIP12.
Among the 34 additional genes involved in the deletion in subject 38, 21 were protein coding and 5 were OMIM morbid genes (COL4A3, COL4A4, MMF, SLC19A3 and TM4SF20). Again, TRIP12 was the only OMIM morbid gene involved in the deletion with a high pLI score (pLI = 1). Subject 34 showed severe intellectual disability with limited speech at 19 years old, behavioral problems, and marked facial dysmorphism. Subject 38 was in the milder end of the clinical spectrum with mild intellectual disability, speech in short sentences at 4 years old and only subtle facial dysmorphism (Fig. 3). Based on the above findings, the phenotype could likely be fully attributed to TRIP12 loss of function in both, but modification of other involved genes cannot be excluded.
Discussion
We report a cohort of 38 individuals with (presumably) disease causing TRIP12 variants. We further delineate the associated phenotype and introduce a TRIP12 facial gestalt.
TRIP12 functions as an E3 ubiquitin ligase, targeting substrates for ubiquitin-mediated degradation by the S26 proteasome. It contains four separate protein domains. The highly conserved HECT domain is responsible for ubiquitin ligase catalytic activity and is therefore essential for normal TRIP12-mediated degradation and signaling. The WWE and ARM domains are interaction domains, allowing binding to different substrates and other protein-protein interactions. The IDR region has a more structural role being involved in interaction stabilization and allows binding of TRIP12 to chromatin. Proteomics analysis led to the establishment of a TRIP12-interacting protein-protein network, consisting of 76 different binding partners mainly involved in the ubiquitination/deubiquitination system on the one hand and transcriptional regulation on the other [10]. The role of TRIP12 in disease was first established in vivo in mice by showing embryonic lethality in homozygous Trip12 mutants, hence demonstrating its crucial role in mouse embryogenesis [20]. In humans, TRIP12 has been linked to cancer with somatic alterations in 1–2% of cancer patients (most frequent alterations in endometrial carcinoma and colon adenocarcinoma) [10] and has been well established as a disease-causing gene for the Clark–Baraitser neurodevelopmental syndrome (OMIM #617752).
Through a European Reference Network ITHACA call, we collected 38 individuals (one previously published) reporting 35 different TRIP12 variants, including 15 frameshift variants, 6 nonsense variants, 5 missense variants, 3 splice variants, 4 intragenic deletions, and two larger deletions disrupting TRIP12. Variants were distributed throughout the entire gene and slightly overrepresented in the WWE protein domain responsible for substrate interaction. Most of these alterations led to a certain or presumed premature stop codon (29/35), subsequently leading to nonsense-mediated decay. A smaller proportion of variants were not premature stop codon variants (6/35). These were enriched in the different protein domains and might have a different biological effect, as was supported by the more severe phenotypic presentation in these subjects. However, as this remains purely hypothetical, further studies will be needed to investigate the functional effects of these variants.
Review of this large cohort allows for thorough characterization of phenotypic traits. Intellectual disability was present in all individuals, marked by language impairment and associated with ASD in 45%. Cognitive performance was mostly in the moderate range, but varied greatly amongst subjects (IQ 40–79). Next, we further confirm obesity, present in half of the individuals, as a cardinal feature of the phenotype. This might be related to hyperphagia, already starting at a young age. Seizures occurred in about 15% and were refractory in the individuals with a missense variant in our cohort. Familial occurrence was described in two independent families and caused, consistent with previous findings, a less severe presentation.
Interestingly, in the adult and more aged individuals, we observed a risk of early cognitive decline as well as hormonal deficits with thyroid dysfunction and osteoporosis. Whether these features are part of the aging TRIP12 phenotype will have to be confirmed by future longitudinal data. Of importance for patient counseling, no elevated risk of cancer or other major health problems were seen at a later age.
We propose a recognizable TRIP12 facial gestalt, marked by characteristic deep-set eyes, a broad nasal tip, a wide mouth and low-set ears with large earlobes. GestaltMatcher facial analysis confirmed these findings, suggesting a distinct TRIP12 facial gestalt.
In conclusion, we further establish the TRIP12-associated neurodevelopmental phenotype and expand the clinical spectrum by reporting the largest cohort to date consisting of 38 individuals with 35 different variants. We elaborate on the aging phenotype and provide a TRIP12 facial gestalt. These findings contribute to a further delineation of this rare disorder, hence aiding in patient counseling and support.
Data availability
All data generated or analysed during this study are either included in this published article or available from the corresponding author on reasonable request. All variants were uploaded in the DECIPHER database (DECIPHER Patient ID 489845-489882) [13].
References
Clark RD, Baraitser M. A new X-linked mental retardation syndrome. Am J Med Genet. 1987;26:13–5.
Louie RJ, Friez MJ, Skinner C, Baraitser M, Clark RD, Schwartz CE, et al. Clark-Baraitser syndrome is associated with a nonsense alteration in the autosomal gene TRIP12. Am J Med Genet Part A U S. 2020;182:595–6.
Lelieveld SH, Reijnders MRF, Pfundt R, Yntema HG, Kamsteeg E-J, de Vries P, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–6.
O’Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014;515:216–21.
Bramswig NC, Lüdecke H-J, Pettersson M, Albrecht B, Bernier RA, Cremer K, et al. Identification of new TRIP12 variants and detailed clinical evaluation of individuals with non-syndromic intellectual disability with or without autism. Hum Genet. 2017;136:179–92.
Zhang J, Gambin T, Yuan B, Szafranski P, Rosenfeld JA, Al Balwi M, et al. Haploinsufficiency of the E3 ubiquitin-protein ligase gene TRIP12 causes intellectual disability with or without autism spectrum disorders, speech delay, and dysmorphic features. Hum Genet. 2017;136:377–86.
Donoghue T, Garrity L, Ziolkowski A, McPhillips M, Buckman M, Goel H. Novel de novo TRIP12 mutation reveals variable phenotypic presentation while emphasizing core features of TRIP12 variations. Am J Med Genet Part A U S. 2020;182:1801–6.
George AJ, Hoffiz YC, Charles AJ, Zhu Y, Mabb AM. A comprehensive atlas of E3 ubiquitin ligase mutations in neurological disorders. Front Genet. 2018;9:29.
Brunet M, Vargas C, Larrieu D, Torrisani J, Dufresne M. E3 ubiquitin ligase TRIP12: regulation, structure, and physiopathological functions. Int J Mol Sci. 2020;21:8515.
Hsieh T-C, Bar-Haim A, Moosa S, Ehmke N, Gripp KW, Pantel JT, et al. GestaltMatcher facilitates rare disease matching using facial phenotype descriptors. Nat Genet. 2022;54:349–57.
Zhang Z, Song Y.Qi H, Age progression/regression by conditional adversarial autoencoder. IEEE Conf Comput Vis Pattern Recognit, CVPR 2017. 2017;4352–60.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84:524–33.
Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–74.
Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–94.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–23.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–24.
Laan L Van Der, Rooney K, Alders M, Relator R, Mcconkey H, Kerkhof J, et al. Episignature mapping of TRIP12 provides functional insight into Clark–Baraitser syndrome. Int J Mol Sci. 2022;23:13664.
Styne DM, Arslanian SA, Connor EL, Farooqi IS, Murad MH, Silverstein JH, et al. Pediatric obesity-assessment, treatment, and prevention: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2017;102:709–57.
Kajiro M, Tsuchiya M, Kawabe Y-I, Furumai R, Iwasaki N, Hayashi Y, et al. The E3 ubiquitin ligase activity of Trip12 is essential for mouse embryogenesis. PLoS One. 2011;6:e25871.
Acknowledgements
The authors would like to thank the patients and caregivers for their participation in this study. We are grateful to Caterina Lo Rizzo and Francesca Ariani for their contribution.
Funding
HVE is supported by a Senior Clinical Investigator fellowship of the Fonds voor Wetenschappelijk Onderzoek (FWO) Flanders. Almost all authors are members of the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability ERN-ITHACA [EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516]. K.Õ is supported by grant from the Estonian Research Council (grant PRG471). The Broad Center for Mendelian Genomics (UM1 HG008900) is funded by the National Human Genome Research Institute with supplemental funding provided by the National Heart, Lung, and Blood Institute under the Trans-Omics for Precision Medicine (TOPMed) program and the National Eye Institute.
Author information
Authors and Affiliations
Contributions
All authors were involved in the collection of clinical information, the genetic analyses/data interpretation and/or the preparation of the manuscript and all approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was conducted according to the UZ Leuven ethical commission guidelines. Appropriate informed consent was obtained for each affected individual, in accordance with the respective local ethical committee. Written informed consent for the publication of pictures was obtained for 21 individuals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aerden, M., Denommé-Pichon, AS., Bonneau, D. et al. The neurodevelopmental and facial phenotype in individuals with a TRIP12 variant. Eur J Hum Genet 31, 461–468 (2023). https://doi.org/10.1038/s41431-023-01307-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01307-x
This article is cited by
-
A Comprehensive Review of Syndromic Forms of Obesity: Genetic Etiology, Clinical Features and Molecular Diagnosis
Current Obesity Reports (2024)
-
April, again
European Journal of Human Genetics (2023)
