
Abstract
Introduction
It is important to confirm the effectiveness and tolerability of disease-modifying treatments for relapsing–remitting multiple sclerosis (RRMS) in real-world treatment settings. This prospective observational cohort study (VIRGILE) was performed at the request of the French health authorities. The primary objective was to evaluate the effectiveness of fingolimod 0.5 mg in reducing the annualised relapse rate (ARR) in patients with RRMS.
Methods
Participating neurologists enrolled all adult patients with RRMS starting fingolimod treatment between 2014 and 2016, who were followed for 3 years. Follow-up consultations took place at the investigator’s discretion. The primary outcome measure was the change in ARR at month 24 after fingolimod initiation. Relapses and adverse events were documented at each consultation; disability assessment (EDSS) and magnetic resonance imagery were performed at the investigator’s discretion.
Results
Of 1055 eligible patients, 633 patients were assessable at month 36; 405 (64.0%) were treated continuously with fingolimod for 3 years. The ARR decreased from 0.92 ± 0.92 at inclusion to 0.31 ± 0.51 at month 24, a significant reduction of 0.58 [95% CI − 0.51 to − 0.65] relapses/year (p < 0.001). Since starting fingolimod, 461 patients (60.9%) remained relapse-free at month 24 and 366 patients (55.5%) at month 36. In multivariate analysis, no previous disease-modifying treatment, number of relapses in the previous year and lower EDSS score at inclusion were associated with a greater on-treatment reduction in ARR. The mean EDSS score remained stable over the course of the study. Sixty-one out of 289 (21.1%) patients presented new radiological signs of disease activity. Treatment-related serious adverse events were lymphopenia (N = 21), bradycardia (N = 19), elevated transaminases (N = 9) and macular oedema (N = 9).
Conclusions
The effectiveness and tolerability of fingolimod in everyday clinical practice are consistent with findings of previous phase III studies. Our study highlights the utility of fingolimod for the long-term management of patients with multiple sclerosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Little information is available on the effectiveness and tolerability of fingolimod in patients with multiple sclerosis in real-world clinical practice. |
A sample of over 1000 patients starting fingolimod treatment was followed for up to 5 years. |
Effectiveness in this setting was similar to that observed in randomised clinical trials. |
No unanticipated safety issues were identified. |
Introduction
The last decade has seen the introduction of many new disease-modifying treatments (DMTs) for multiple sclerosis (MS). Fingolimod, a sphingosine-1-phosphate receptor modulator [1,2,3], was the first orally administered DMT to be approved (in 2011 in Europe). The efficacy and safety of fingolimod to reduce the risk of relapse were demonstrated in two large phase III randomised clinical trials comparing fingolimod to placebo (FREEDOMS study [4]) and to intramuscular interferon-β1a (TRANSFORMS study [5]). On the basis of these studies, fingolimod (capsule 0.5 mg/day) was approved for the treatment of MS in Europe in 2011.
As for all new therapies, it is important to confirm the effectiveness and tolerability observed in phase III interventional trials in real-world settings, where patients may be in worse overall health and receive less intensive follow-up. This issue is particularly relevant in conditions such as MS, which evolve over time and where many treatment choices now exist, leading to frequent treatment discontinuation and switching [6,7,8]. In the case of fingolimod, prospective real-world data are relatively limited [9]. Some real-world studies have focussed on single outcomes such as treatment discontinuation [10, 11], others have used insurance claims databases, which contain limited information on clinical outcomes [12, 13], whilst others have included relatively low numbers of subjects (fewer than 350) [14,15,16]. The most comprehensive data available comes from the PANGAEA study, a large cohort of more than 4000 patients in Germany, and followed up for 5 years. The findings of this study suggest that sustained effectiveness and manageable tolerability can be achieved with fingolimod in the real-world setting [17].
During the licensing process for fingolimod in France in 2011, the French health authorities (Haute Autorité de Santé; HAS) requested that a post-reimbursement pharmacoepidemiological drug utilisation study be conducted to assess the use and impact of fingolimod in the treatment of highly active forms of relapsing–remitting MS (RRMS) in a real-world treatment setting in France. Objectives were to assess the effect of fingolimod 0.5 mg on relapse activity, disability progression and quality of life. The study also provided an opportunity to describe the characteristics of patients treated with fingolimod and natalizumab, and to estimate the impact of fingolimod treatment on healthcare resource utilisation in both groups. Similar drug utilisation studies have been requested and performed for other DMTs for MS, such as glatiramer acetate (GA) [8] and natalizumab [18, 19], and are also currently underway for other recently approved treatments.
The primary evaluation criterion of VIRGILE was the change in annualised relapse rate (ARR) compared to baseline following 2 years of treatment with fingolimod. Secondary study objectives were to describe the profile of patients treated with fingolimod in France, to evaluate changes in disability, ARR and radiological markers of MS disease activity, to evaluate changes in health-related quality of life (HRQoL), to document treatment persistence, discontinuation and treatment switches, to document safety and to document healthcare resource utilisation. The data on resource utilisation will be presented elsewhere.
Methods
VIRGILE is a non-interventional, multicentre, prospective, observational study conducted in France by neurologists treating patients with MS. The design and implementation of the study were overseen by an independent multidisciplinary scientific committee. The study was initiated in collaboration with the French MS Observatory (Observatoire Français de la Sclérose en Plaques; OFSEP). Eligible patients were enrolled between January 2014 and February 2016. At the request of the French health authorities, a parallel group of patients initiating natalizumab was also included. The design of the study followed the guidelines of the French health authorities for drug utilisation studies [20] and the protocol was validated by the HAS in 2012.
The study was closed after the last patient enrolled had completed 3 years of follow-up (February 2019) with fingolimod. Patients in the fingolimod group who completed 3 years of follow-up could, if they chose, continue in the study until it closed (up to 2 years of additional follow-up). The follow-up duration for a given patient could thus range from 3 to 5 years. No specific study-related procedures were required by the study protocol and participation in the study did not influence the care or management of patients. Follow-up patient consultations after the inclusion visit were planned according to the standard practice of the participating neurologist. This was anticipated to be every 6 months, since this is the recommended follow-up interval in the prescribing information for this treatment [21], and the usual duration of a single fingolimod prescription in France. A window of ± 3 months was considered acceptable.
Participants
Postal invitations to participate in the study were sent to all hospital-based neurologists in France treating patients with MS identified from a national list. Community-based neurologists were only eligible if they also saw patients in a hospital or clinic, since the prescribing information for fingolimod specifies that this medication should be initially prescribed in hospitals only. These 1361 physicians correspond to around half of all practising neurologists in France. The invitation included an answer form and a prepaid return envelope. In the case of non-response, invitees were contacted by post, electronic mail or telephone until the target quota of 300 participating neurologists was reached.
All patients with a confirmed diagnosis of MS according to the revised McDonald criteria [22] and for whom a first prescription of fingolimod had been made in its approved indication at the inclusion consultation were eligible for the study. The approved indication for fingolimod in France is ‘the treatment of patients with highly active, relapsing remitting multiple sclerosis (RRMS) despite a full and adequate course of treatment with at least one DMT or with rapidly-evolving severe RRMS defined by two or more disabling relapses in 1 year, and with disease activity visible on magnetic resonance imagery (MRI)’ [21]. Patients were required to agree to study personnel having access to their medical files and to statistical analysis of their personal medical data. Otherwise, eligibility criteria were broad to ensure the representativeness of the study cohort. The only exclusion criteria were current treatment with fingolimod or any other DMT at the inclusion visit and participation in an interventional clinical trial.
Over the 2-year inclusion period, each participating neurologist was expected to invite sequentially all patients fulfilling the eligibility criteria to participate in the study, up to a maximum of 48 patients by centre. Patients starting treatment in the context of a clinical trial were excluded.
An independent cohort of patients starting treatment with natalizumab was also recruited in the same way, using the same eligibility criterion. The approved indication for natalizumab in France is ‘treatment of adults with highly active RRMS with highly active disease despite a full and adequate course of treatment with at least one DMT or with rapidly evolving severe RRMS defined by ≥ 2 disabling relapses in 1 year, and with ≥ 1 gadolinium enhancing lesions on brain MRI, or a significant increase in T2 lesion load as compared to a previous recent MRI’.
Data Collection
Data could be collected in one of two ways. Participating centres who were members of OFSEP could use the web-based interface of the European Database for Multiple Sclerosis (EDMUS) which is used routinely in these centres for collecting patient data in MS [23]. Other centres not participating in OFSEP were provided with an electronic or paper case report form specific for the study.
At the inclusion visit, data was collected on demographics, general and MS-specific medical history. At all visits, disability was evaluated using the Expanded Disability Status Scale (EDSS) [24] and any MS relapses since the previous visit were documented. Magnetic resonance imagery (MRI), usually yearly brain MRI, was performed according to the standard practice of the physician, who could choose the MRI sequence used and the timing of the investigation. Treatment data included initiation, switch or discontinuation of any MS-specific medication, and the reason for any change, and any monitoring procedures required for fingolimod. The occurrence of adverse events (AEs) was documented following non-directed questioning from the neurologist at each study visit, and each AE was qualified in terms of seriousness, severity, duration and causality. At each study visit, each patient was provided with two HRQoL questionnaires, the MusiQoL [25] and the EQ-5D-3L [26], as well as a questionnaire on healthcare resource utilisation.
Study Outcomes
The primary effectiveness outcome in the study was the change in ARR after 24 months of treatment with fingolimod, determined both as an absolute and as a relative value. Secondary effectiveness outcomes included other relapse-related variables, disability measures and MRI measures. The ARR was also determined at 12 and 36 months and the proportion of patients who had been relapse-free since inclusion was documented. The variation in mean and median EDSS scores between study visits was assessed. Disability was considered to have progressed if the EDSS score had increased by at least one point (or 0.5 points if the previous score was 5.5 or higher) since the previous visit and if this increase was confirmed at 6 months. Imaging outcomes included the number of new or enlarged T2 lesions accrued since inclusion and the number of gadolinium-enhancing lesions visible at each evaluation. Safety outcomes consisted of adverse event reporting, electrocardiogram (ECG) monitoring and clinical laboratory tests.
Statistical Analysis
Data were analysed for three patient populations. The safety population included all patients who were treated with fingolimod during the study, irrespective of eligibility status. The full analysis set (FAS) consisted of all eligible patients included in the study and given a prescription for fingolimod, irrespective of what happened to them during the study. The continuous treatment set (CTS) consisted of those members of the FAS who were still in the study and taking fingolimod at the end of the 3-year observation period. In addition, a sensitivity analysis of the key effectiveness variables was performed in the subset of patients treated in strict accordance with the prescribing information for fingolimod (PI subset). Missing data due to incomplete attendance at the different study visits were not replaced, and outcomes are analysed on an observed cases basis.
The sample size was determined a priori in order to fulfil two goals. The first was to be able to detect a significant change in ARR 2 years after inclusion with a power of 90%, assuming a standard error of 1 and an α-risk of 0.05 (Wilcoxon test). The second goal was to optimise representativeness of the subgroup of patients previously treated by natalizumab and to obtain a sufficiently large subgroup (at least 60 patients) who had never previously received a DMT (treatment-naïve patients). These hypotheses were based on relapse rates and treatment patterns observed in a previous observational study of fingolimod in France (GRACE) [27]. To meet the first goal, 456 patients would have been sufficient, but the second goal required 1200 patients to be included, assuming that only 5% of patients starting fingolimod are treatment-naïve.
The primary outcome measure (change in ARR after 24 months of treatment) was compared between the inclusion visit and the month 24 study visit using a paired Wilcoxon test. For the relative change in ARR, patients who did not experience a relapse in the year preceding inclusion were excluded from the analysis. Secondary outcome measures are presented descriptively.
The potential association of certain variables with an on-treatment change in ARR at 24 months was evaluated using multivariate logistic regression analysis. The following variables were selected a priori: age (by quartile), gender, disease duration (time since first symptoms in four classes: 0–3 years, 4–7 years, 8–10 years and more than 10 years), number of previous DMTs (five classes: none, one, two, three and four or more), reason for switching to fingolimod (efficacy, tolerability or other), type of DMT most recently used (four classes: DMT-naïve, interferon-β or GA, natalizumab and other DMT), relapses in the year preceding inclusion (absent or present) and EDSS score at inclusion (at most 3 or more than 3). Association of these variables with the absolute change in ARR at month 24 was tested first in a univariate analysis for individual variables. In the second step, all significant variables identified in the first step were tested simultaneously in a backward stepwise multivariate regression analysis, in which variables with no significant association with change in ARR at a probability threshold of 5% were eliminated after each round.
Continuous data are presented as median or mean values with their 95% confidence values or standard errors and categorical values as frequency counts with percentages. All analyses were performed on SAS software version 9.2 (SAS Institute, Cary, USA).
Ethics
The study was conducted in accordance with the Declaration of Helsinki and with all other relevant international and French legislation and followed Good Clinical Practice. The protocol was submitted for approval to the CPP (Comité de Protection des Personnes; ethics committee), and the opinion of the CPP was transmitted to the French Safety of Medicines Agency and to the CCTIRS (Comité Consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé), as required under French law for observational studies. Approval (No. 13.307 dated 22 May 2013) was at a national level and covered all participating centres. Prior to inclusion, each eligible patient was provided with a study information leaflet and was given the opportunity not to participate in the study. Patient consent to the collection, analysis and potential monitoring of the data required for the study was given and documented in the study database. Patient enrolled through OFSEP also signed the OFSEP consent form. All patient data was anonymised before entry into the database. The study protocol was submitted to the French Data Protection Agency (Commission Nationale de l’Informatique et des Libertés; CNIL) for comment before initiation of the study.
Results
Participants
Of 1361 neurologists contacted, 419 (30.8%) agreed to participate, 332 (24.4%) signed the study contract and received the study materials and 188 (13.8%) in 123 individual centres recruited at least one patient. Of these active neurologists, 114 (60.6%) used the EDMUS interface for data collection and 74 (39.4%) used the stand-alone case report form.
A total of 1099 patients were enrolled and prescribed fingolimod and these constituted the safety population. Forty-four enrolled patients (4.0%) were excluded owing to infringements of the eligibility criteria, most frequently because the treatment was not initiated at the inclusion visit, the date of treatment initiation was not known or because fingolimod had been prescribed previously. The remaining 1055 eligible patients constituted the FAS.
Patient Disposition
A patient flow diagram is provided in Fig. 1. Over the 3-year follow-up period, 135 of the 1055 patients in the FAS (12.8%) left the study before the end of the 3-year follow-up period (Fig. 1). The most frequent reasons for leaving the study were a change in the treating neurologist, loss to follow-up of the patient and decision of the patient to leave the study. In addition, three patients died (by the month 6, month 12 and month 30 study visits). Since follow-up visits were decided at the discretion of the investigator and were not obligatory, not all patients remaining in the study attended each 6-monthly study visit. A total of 663 patients made the month 36 study visit and could be assessed at this time, of whom 405 (61.1%) had been treated with fingolimod continuously and these latter patients constituted the CTS.

Patient disposition. FAS full analysis set, CTS continuous treatment set, PI prescribing information compliant, RRMS relapsing–remitting multiple sclerosis. Percentages are calculated with respect to the number of patients enrolled in each case. Multiple reasons can be provided for fingolimod discontinuation and, for this reason, the response modalities are not mutually exclusive
In addition, 342 patients were enrolled and prescribed natalizumab and these constituted the natalizumab safety population. Twelve enrolled patients (4.0%) were excluded because of infringements of the eligibility criteria. The remaining 330 eligible patients constituted the natalizumab FAS. Thirty-three patients left the study before the end of the 3-year follow-up period. A total of 241 patients made the month 36 study visit and could be assessed at this time.
Patient Characteristics
The characteristics of the 1055 patients initiating fingolimod who constituted the FAS are presented in Table 1. Around three-quarters of the cohort were women and the mean age was 41 years. Over 90% of patients presented with RRMS and the mean disease duration was around 10 years. Overall, 37.5% of patients had been free of relapses in the previous year. The majority of patients who had been relapse-free in the 12 months preceding inclusion (N = 260; 65.8%) were patients who had been switched from natalizumab (76.7% of previous natalizumab patients). In contrast, only nine (8.1%) of treatment-naïve patients and 92 (19.9%) of patients previously treated with interferon-β or GA were relapse-free in the previous 12 months. The EDSS score at inclusion was at most 3 in 65.2% of patients.
The characteristics of patients in the natalizumab arm are presented in Table 1. Compared to the patients initiating fingolimod treatment, these patients were on average younger, with a shorter disease duration, but had more active disease in terms of relapses prior to inclusion and had accrued more disability. In addition, they were more frequently men and treatment-naïve.
Ten percent of patients in the fingolimod arm (N = 110) were treatment-naïve. For the remaining patients who were switched from another DMT, the previous treatment was most frequently natalizumab or an interferon-β (N = 339 and N = 329 respectively; Fig. 2). The most frequent reason for discontinuing the previous treatment was an inadequate treatment response (N = 441; 42.3%), planned discontinuation, principally in the case of patients switched from natalizumab (N = 223; 21.4%) or the occurrence of an AE (N = 118; 11.3%). In the natalizumab arm, 22% of patients (N = 73) were treatment-naïve and a similar proportion switched from interferon-β (N = 77; 23.3%). Around 15% of patients were switched for each of GA (N = 47), fingolimod (N = 59) and dimethylfumarate (N = 44).

Previous disease-modifying treatments (FAS). Data correspond to the last disease-modifying treatment used before inclusion in the study. DMF dimethylfumarate, Exp experimental therapy, FGM fingolimod, GA glatiramer acetate, IFN-β interferon-β, NTZ natalizumab, OCR ocrelizumab, TFL teriflunomide; 1Azathioprine, cyclophosphamide, methotrexate or mycophenolate mofetil. 2Laquinimod, siponimod, daclizumab, high-dose vitamin D or zaurategrast
A comparison of the baseline characteristics of the analysis population according to the previous DMT treatment is provided in Supplementary Material Table 1.1. Overall, 1018 (96.5%) patients had undergone MRI in the year preceding inclusion, and 620 (62.4%) within the preceding 3 months. At the time of inclusion, 748 of the 913 patients evaluated (81.9%) presented at least nine lesions on T2-weighted images, and 322 out of 960 (33.5%) presented one or more gadolinium-enhancing lesion on T1-weighted images.
Fingolimod was prescribed in accordance with its prescribing information in 593 patients (56.2%). Most of these patients (N = 359; 94.3%) were switched from another DMT because of persistent disease activity, the remaining 34 patients being treatment-naïve with rapidly evolving, severe RRMS. These constituted the prescribing information compatible (PI) subset. For the remaining 462 patients, it was not explicitly specified that the treated patient presented with a relapsing–remitting form of MS in 155 cases (33.5%). In addition, 243 patients were switched from another DMT to fingolimod even though their MS was well controlled on previous therapy, and 31 patients who were treatment-naïve did not fulfil criteria for rapidly evolving, severe RRMS. Information on patient characteristics and outcomes in the PI subset is provided in Supplementary Material 2.
Primary Effectiveness Outcome: Change in Annualised Relapse Rate at Month 24
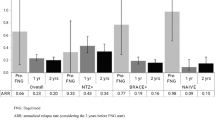
The change in ARR between inclusion and month 24 could be evaluated in 758 patients of the FAS who attended this study visit. In these patients, the mean ARR decreased from 0.92 ± 0.92 in the year preceding inclusion to 0.31 ± 0.51 at month 24 (Fig. 3). The mean absolute change in ARR between the two periods was − 0.58 [95% CI − 0.51 to − 0.65], a statistically significant reduction (p < 0.001; paired Wilcoxon test).

Annual relapse rate over the course of the study (FAS and CTS sets). Data are presented as mean values with their 95% confidence intervals. Light columns, full analysis set; dark columns, continuous treatment set. The numbers below the columns (N) indicate the number of patients available for analysis at each time point
Relative changes in ARR could only be determined for the subgroup of 659 patients who experienced relapses in the year before inclusion, 469 of whom were assessable at month 24. In this subgroup, the absolute reduction in ARR at month 24 was 1.11 ± 0.92 (p < 0.001; Wilcoxon paired rank test), corresponding to a mean relative reduction of 76.3% ± 38.6%. For the remaining subset of 395 patients who were relapse-free in the year prior to inclusion, 288 were assessable at month 24. In this subgroup, the absolute increase in ARR at month 24 was 0.29 [0.23–0.34]. A comparison of relapse-related outcome variables in the analysis population according to the previous DMT treatment is provided in Supplementary Material Table 1.2.
In the CTS, the mean ARR decreased from 0.88 ± 0.89 at inclusion to 0.17 ± 0.36 at month 24. The mean absolute change in ARR at month 24 was − 0.69 ± 0.92 (Fig. 3). For the 216 members of the CTS with at least one relapse in the year preceding inclusion, the relative change in ARR at month 24 was − 84.9% ± 31.0% (absolute change − 1.22 ± 0.77 relapses/year).
In the PI subset of patients who received fingolimod in accordance with its approved indications, the mean ARR decreased from 1.29 ± 0.83 at inclusion to 0.33 ± 0.53 at month 24. The mean absolute and relative changes in ARR at month 24 in this subgroup were − 0.92 ± 0.90 and − 74.8% ± 38.9% respectively. Further information on the effectiveness of fingolimod in this patient subgroup is provided in Supplementary Material 2.
In the natalizumab cohort, the mean ARR decreased from 1.59 ± 1.01 at inclusion to 0.29 ± 0.49 at month 24. The mean absolute change in ARR at month 24 was − 1.32 ± 1.01 (Fig. 3). For the 294 patients with at least one relapse in the year preceding inclusion, the mean relative change in ARR at month 24 was − 82.6% ± 32.7% (absolute change − 1.53 ± 0.89 relapses/year).
Secondary Effectiveness Outcome
Relapses
The decrease in ARR was maintained at month 36 (mean change from baseline − 0.63 ± 0.95). The ARR for all patients evaluable at each time point is presented in Fig. 3. At month 24, 461 patients out of the 757 evaluated at this time point (60.9%) had been relapse-free since starting fingolimod. At the end of the 3-year study period, 366 patients (55.5%) in the fingolimod FAS had been relapse-free throughout.
In the natalizumab cohort, the mean ARR at month 36 was 0.26 ± 0.41, corresponding to a mean absolute change from baseline of − 1.38 ± 0.98. The proportion of patients in this cohort who remained relapse-free was 63.6% at month 24 (N = 175) and 55.6% at month 36 (N = 134).
Disability
In the FAS, mean and median EDSS scores increased marginally over the follow-up period (mean 2.6 at month 0 and 2.9 at month 36; median 2.0 at month 0 and 2.5 at month 36). This difference, although modest, was statistically significant (p < 0.05, mixed effects model). The interquartile range remained between 1 and 4 for all visits during the 36-month study period (Fig. 4). In the CTS, the mean EDSS evolved from 2.5 ± 1.7 (median 2, IQR 1–3.5) at inclusion for 287 evaluable EDSS to 2.6 ± 1.9 (median 2, IQR 1–4) at month 36 for 315 evaluable EDSS.

EDSS disability score over the course of the study (FAS set). Data are presented as a box and whisker plot. The white circles represent the mean values (with figure above), the white bars represent the median values, the grey boxes represent the interquartile ranges and the whiskers represent the extreme (minimum and maximum) values. The numbers below the columns indicate the number of patients available for analysis at each time point
In the natalizumab cohort, the mean EDSS score was 3.2 ± 1.88 at inclusion, 2.9 ± 2.0 at month 24 and 3.1 ± 2.1 at month 36.
MRI Activity
Magnetic resonance imaging outcome could only be assessed in patients who had undergone MRI in the 3 months to the inclusion visit and at least once during the 3-year follow-up, and for whom both evaluations included both gadolinium-enhanced T1-weighted and T2-weighted sequences.
Of the 663 patients in the FAS who attended the month 36 study visit, 606 (91.4%) had undergone at least one MRI examination since inclusion, 362 of whom (59.7%) had also been evaluated prior to inclusion. In these 362 patients, the evaluation included both sequences for 289 patients. Radiological signs of disease activity during the study could thus be determined in this subgroup. No new radiological signs of disease activity were observed in 228 patients (78.9%). The remaining 61 patients (21.1%) presented either an increase in the number or volume of T2 lesions since inclusion, the appearance of at least one new gadolinium-enhancing T1 lesion, or both (Fig. 5).

Radiological disease activity over the course of the study. FAS full analysis set, CTS continuous treatment set
In the CTS, 367 patients (90.6%) had undergone at least one MRI examination since inclusion, of whom 224 had also been evaluated in the 3 months preceding inclusion; 178 could be analysed as they were assessed with both MRI sequences. No signs of disease activity following initiation of fingolimod treatment were observed in 157 patients (88.2%) (Fig. 5).
In the natalizumab cohort, MRI outcome was evaluable in 126 patients in the FAS population (38.8%). Of these patients, 111 (88.1%) presented no new radiological signs of disease activity during the follow-up period (Fig. 5).
Variables Associated with On-Treatment Change in Relapse Rate
Of the variables evaluated for an association with change in ARR at month 24 in the FAS, significant associations were observed for time since first symptoms (larger change in patients with more recent disease), number of previous DMTs (larger change in patients with fewer previous treatment), most recent DMT used (largest change for treatment-naïve patients, smallest change for patients switching from natalizumab), reason for starting fingolimod (largest change for treatment-naïve patients, smallest change for patients switching from previous DMT for ‘other reasons’), relapses in the previous year (largest change for patients with relapses) and EDSS score at inclusion (largest change for patients with EDSS no greater than 3 at inclusion) (Table 2). In the multivariate analysis, only the most recent DMT used, relapses in the previous year and EDSS score at inclusion were retained (Table 2). A similar analysis was performed in the CTS, and identified the same variables associated with change in ARR in the univariate analysis. In the multivariate analysis, the absence of relapses in the previous year and EDSS score greater than 3.0 at inclusion, as well as the reason for starting fingolimod were retained.
Treatment
Over the entire study period, fingolimod was definitively discontinued in 418 patients of the safety population (38.0%), of whom 119 patients (10.8%) discontinued in the first 6 months and 380 patients before the end of the 3-year principal study duration (34.6%). The mean treatment duration before fingolimod discontinuation was 17.1 ± 13.4 months (median 14 months). The principal reasons for fingolimod discontinuation were inadequate efficacy (N = 148; 35.7% of discontinuations), poor tolerability (N = 141; 34.0%) and patient request (N = 45; 10.8%). In addition, 37 patients (8.9%) stopped fingolimod because they wished to become pregnant. Following fingolimod discontinuation, 103 patients (24.6% of discontinuations) received no further documented treatment, whereas 274 (65.6%) were switched to another DMT (data was missing for the remaining 41 patients). The most frequent first switch choices were natalizumab (N = 65; 23.7% of switches), rituximab (N = 52; 19.0%) and dimethylfumarate (N = 41; 15.0%), although multiple sequential treatment changes were observed in many patients. Eleven patients subsequently reverted to fingolimod after having switched to another DMT. In addition to these permanent discontinuations, 31 patients temporarily discontinued fingolimod for a mean duration of 20 days, and a further 39 patients reduced the dose of fingolimod during the course of the study, in most cases (23 patients) from 0.5 mg once a day to 0.5 mg every other day.
Quality of Life
Quality of life was assessed in the 514 patients of the FAS who returned a patient questionnaire at inclusion and at 36 months (or at the last study visit, if the patient left the study prematurely). The mean MusiQoL score remained stable from 67.8 ± 15.9 at inclusion to 66.9 ± 17.1 at the last study visit. The mean change between the two evaluations was − 0.95 ± 14.0. The most impacted scale dimensions were rejection, relationships with the healthcare system and relationships with family. No differences in any of the dimension scores were observed between inclusion and the last study visit. Quality of life scores were analysed separately in treatment-naïve patients and in patients switched from a previous DMT. No difference was observed between these two groups either in mean MusiQoL scores at inclusion (68.1 ± 16.0 versus 67.7 ± 15.9 respectively) or in mean changes in score on treatment (0.51 ± 12.2 versus − 1.14 ± 14.2 respectively). In the natalizumab cohort, the mean MusiQoL score was 64.8 ± 16.9 at inclusion and 67.6 ± 17.5 at the last study visit (mean change 3.5 ± 16.3), a non-significant difference.
Similarly, the mean global EQ-5D scale remained stable between inclusion (0.66 ± 0.26) and the last study visit (0.66 ± 0.26), with a mean change between the two visits of 0.00 ± 0.21 points. Again, no differences were observed between treatment-naïve patients and those previously exposed to other DMTs (0.66 ± 0.25 and 0.66 ± 0.26 respectively for scores at inclusion and − 0.02 ± 0.19 and 0.00 ± 0.21 for on-treatment change). In the natalizumab cohort, the mean EQ-5D score was 0.62 ± 0.29 at inclusion and 0.69 ± 0.28 at the last study visit (mean change 0.08 ± 0.25), a non-significant difference.
Safety
In the safety population of 1099 patients who received fingolimod, 662 (60.2%) presented at least one AE following treatment initiation (Table 3). These were considered treatment-related in 509 patients (47.3%) and necessitated treatment discontinuation in 125 cases (11.7%). The most frequently reported AEs were lymphopenia, increased transaminases, leucopenia, increased γ-glutamyltransferase and asthenia. Concerning AEs of specific interest for fingolimod, bradycardia was reported in 40 patients, hypertension in 30, atrioventricular block in six, macular oedema in 14, herpes zoster in 12 and basal cell carcinoma in four patients.
Serious AEs (SAEs) were reported in 162 patients (14.8%). The most frequently reported SAEs were lymphopenia (22 patients), bradycardia (21 patients), elevated transaminases (9 patients) and macular oedema (9 patients), which were all, with the exception of one case of lymphopenia and two of bradycardia, considered to be related to fingolimod treatment.
As recommended in the prescribing information, all patients starting treatment with fingolimod were monitored for cardiovascular safety at the first dose, with an ECG being performed within the first 6 h in all patients. During the 6-h observation period, bradycardia with a heart rate below 45 bpm was observed in 19 patients (1.8%), which was symptomatic in two patients, and one patient presented a grade III atrioventricular block. The reduction in cardiac frequency reached a nadir within 5 h in 82% of patients. At the end of the 6-h observation period, six patients presented a grade II atrioventricular block, two presented a QTc interval of at least 500 ms and 12 bradycardia with a heart rate below 45 bpm.
In the natalizumab cohort, at least one AE was documented in 180 patients (52.6% of the safety population) (Table 3). These were considered natalizumab-related in 91 patients (28.6%) and led to natalizumab discontinuation in 23 (7.0%). The most frequent individual AEs reported were asthenia (45 patients; 13.2%), headache (36 patients; 10.5%) and nasopharyngitis in 26 patients (7.6%). Serious AEs were reported in 27 patients (8.0%), and these were considered natalizumab-related in seven patients. These seven events were two cases of elevated transaminases, and one case each of bradycardia, breast cancer, progressive multifocal leukoencephalopathy (PML), hepatocellular lesion, rash and pityriasis rosea.
Three cases of PML were reported during the study. Two cases occurred in the fingolimod cohort, the first of these 31 days after starting fingolimod following 4 years’ treatment with natalizumab and the second 28 days after switching from natalizumab which had been prescribed for over 7 years. In the first of these cases, the patient, a 44-year-old JC-virus-positive man, died of pulmonary disease and toxic shock associated with the PML 22 days after being hospitalised. A posteriori, it was found that the PML lesion was already visible on an MRI scan 20 days after starting fingolimod. In the second case of PML, in a 49-year-old woman, neurological symptoms of PML appeared before treatment with fingolimod was started, although the diagnosis was only formally made 3 weeks later on the basis of MRI findings and a positive JC virus screen in a lumbar puncture sample. Following treatment with corticosteroids, the patient recovered with neurological sequelae. In both these cases of PML, fingolimod was not suspected and the infection was attributed to the long previous history of exposure to natalizumab. The third case, again non-fatal, was reported in a patient in the natalizumab cohort who had been treated with this medication for 2 years and 5 months.
Five deaths occurred during the study, including the case of PML discussed above. One of the other deaths was due to lung cancer which occurred 52 days after initiation of fingolimod. The cause of death in a 75-year-old patient with diabetes in the fingolimod cohort, who died 30 months after starting fingolimod and 9 months after discontinuing treatment, was not documented. Neither of these two deaths was considered causally related to fingolimod. One 32-year-old woman died of pulmonary embolism 20 months after initiation of natalizumab; the relationship to treatment was not reported. The final case concerned a 52-year-old woman who died of septic shock associated with suspected pyloric stenosis or volvulus, 4 months after starting natalizumab. A causal relationship with natalizumab was not suspected.
Discussion
The VIRGILE observational study followed 1055 patients starting treatment with fingolimod in France in everyday clinical practice over a 3-year period. Around two-thirds of subjects persisted with fingolimod treatment for the full 3-year period. The primary effectiveness outcome measure was the change in ARR at month 24. At this time, the mean ARR had decreased to 0.31 relapses/year for the full study cohort and to 0.17 relapses/years for the patients taking fingolimod continuously for 3 years, with 56% of patients remaining relapse-free for this period. This decrease was sustained until the end of the follow-up period.
The study has a number of strengths and limitations. The strengths include the relatively large number of participating centres and of patients included. It is estimated that around one-quarter of all patients initiating treatment with fingolimod in France during the enrolment period participated in the VIRGILE study. It thus should provide an accurate picture on how fingolimod is used in clinical practice, avoiding the potential pitfalls of inclusion bias and evaluation bias in smaller cohorts. Secondly, we evaluated not only clinical outcomes but HRQoL, using two different measures. In addition, the study used standard EDMUS software for data collection in approximately 75% of participating centres, which will allow the information to be compared with other data sets collected in the different research programmes initiated by OFSEP.
Limitations principally reflect the observational nature of the study. In particular, there was a large amount of missing data at each study visit, since these visits were not programmed systematically every 6 months as recommended in the prescribing information for fingolimod. In particular, pre- and post-treatment MRI was only performed for one-third of the included patients. In addition, no information is available on patient compliance with the prescribing recommendations for fingolimod or on whether treatment was actually taken. In the absence of a placebo group, any contribution of regression to the mean to the change in clinical outcome over time cannot be assessed. This may occur because patients are generally enrolled following a relapse, so they would have a natural tendency to recover (improve) independently of treatment. On-treatment reductions in ARR were, de facto, not observed for patients without relapses in the year preceding inclusion nor in those switching from natalizumab (these two subgroups of patients overlapped extensively). The issue of regression to the mean is addressed in part by the multivariate analysis of variables associated with the change in ARR. In the CTS, the effect size was similar whatever the reason for switching the previous DMT to fingolimod (persistent disease activity, AEs or ‘other’), suggesting that there is a real treatment effect, over and above any contribution of regression to the mean. Finally, since many enrolled patients had not experienced a relapse in the year preceding inclusion, the relative reduction in ARR could not be calculated for these patients.
Patients treated with fingolimod were in general switched from another DMT (only 10% were treatment-naïve), most frequently following planned discontinuation of natalizumab. In terms of disease activity at inclusion, the patients treated were not very severe, with a median EDSS score of 2 and an ARR in the previous year of less than 1, in spite of having a 10-year (on average) history of MS. The age and gender distributions of these patients were consistent with those of the overall RRMS population in France [28].
Over half of treated patients were treated consistently with the approved indications of fingolimod, the most frequent inconsistency being switches from another DMT to fingolimod even though their MS was well controlled on previous therapy. Two possible explanations can be proposed for the rather high rate of prescription outside the approved indication. The first is that the duration of treatment with natalizumab was limited to 2 years at the time the present study was initiated, and natalizumab treatment is also required to be discontinued in patients who develop anti-JC virus antibodies. For patients who had to stop natalizumab therapy for these reasons, even if their MS was well controlled, fingolimod was the only alternative second-line treatment available for most of the length of the study. Patients switching from natalizumab constituted the majority of patients starting fingolimod outside the approved indication. The second explanation would be that the oral route of administration of fingolimod encouraged patients treated with interferon-β or GA who were averse to self-injection to switch to fingolimod, even if they were well controlled.
Over the 3-year follow-up period, 61.1% of patients had been treated with fingolimod continuously. The rate of treatment discontinuation was higher than that reported in previous real-world studies performed in Sweden [10] and Canada [11]. In addition, the principal reason for stopping fingolimod was inadequate efficacy, whereas in the earlier studies it was AEs. This difference may be explained by the wider range of treatment options available during the VIRGILE study, notably other oral DMTs and anti-CD20 monoclonal antibodies, which encourages switching from fingolimod when the treatment response is suboptimal. Similar relatively high rates of switching from and between fingolimod and dimethylfumarate have been reported in a recent study from the USA [29].
The relatively large sample size made it possible to investigate certain patient variables for association with treatment response, in terms of change in ARR. Relapses in the year before inclusion, less disability at inclusion and no previous DMT treatment were all associated with a better treatment response. A sensitivity analysis was performed in the CTS, in which three variables were retained; ARR in the previous year, EDSS score at baseline and reason for staring fingolimod were identified as predictors of response. Although in this analysis, the ‘reason for starting fingolimod’ variable replaces the ‘previous DMT’ variable in the FAS, both variables had ‘no previous treatment’ as a response modality which, in both data sets, was associated with a better treatment response, so that the two analyses are consistent. These findings can be compared with previous studies of predictors of fingolimod response. For example, in a Spanish observational cohort study [15], which evaluated 211 patients, only baseline EDSS score was associated with the on-treatment ARR in the first 24 months of fingolimod treatment, but the statistical power of that study was relatively low compared to the present VIRGILE study. A recent Italian study with RRMS evaluated predictors of ‘no evidence of disease activity’ (NEDA), time to first relapse and time to confirmed disease progression in a cohort of 339 patients treated for 4 years with fingolimod [30]. Variables associated with time to first relapse were age at disease onset, gadolinium-enhancing lesions at inclusion, ARR in the previous 2 years and natalizumab treatment in the previous year. Other studies have evaluated biological parameters, such as lymphocyte populations, as predictors of fingolimod response [31, 32], but this was not feasible in the present study. Associations between pre-treatment disease activity, baseline disability and treatment history on the one hand and clinical response on the other have been observed previously in long-term observational studies of patients treated with interferon-β [33, 34] or GA [8, 35].
The findings of the VIRGILE study can be compared with those of the phase III clinical trials of fingolimod (FREEDOMS [4] and TRANSFORMS [5]). Persistence rates were much higher in the clinical trials (approx. 85%) than in VIRGILE, consistent with the interventional nature of these studies, and around half of patients included in these trials were previously treatment-naïve. On-treatment ARRs in these studies were 0.18 [4] and 0.16 [5], and the proportions of patients remaining relapse-free were 70% [4] and 83% [5]. As in fingolimod-treated patients in the phase III trials, the EDSS score remained essentially stable in the VIRGILE study. Although in the present study the change in score over the 3-year evaluation period was statistically significant, the change in mean score was not considered clinically relevant.
The findings can also be compared with those of the PANGAEA study [17], the largest real-world study published with fingolimod to date. It should be noted that effectiveness is only described in this study for patients still treated with fingolimod at the time of the follow-up visit, corresponding to the CTS in the VIRGILE study. In the PANGAEA study, fingolimod persistence rate at 36 months was 70%, compared to 56% in VIRGILE. The PANGAEA patients were principally switched to fingolimod from an interferon-β, and thus may explain the higher baseline ARR (1.79 relapses/year) compared to the VIRGILE study. Over the 3-year treatment period, the ARR in PANGAEA was 0.27, 58% of patients were relapse-free and 13% presented confirmed disease progression. Corresponding data in the VIRGILE study (FAS population) were 0.31, 55.5% and 23.4% respectively. The findings of the two studies can thus be considered comparable.
Regarding safety, the study provides useful information on exposure in unselected patients, including those from potentially at-risk groups, who were excluded from the phase II and III clinical trials, such as the elderly and those with certain comorbidities. In addition, the large number of patients included should facilitate the detection of rare events. Nonetheless, the nature of the AEs reported was essentially similar to those reported in the phase III trials [4, 5], and no unexpected AE, in particular no unexpected serious AE, was reported in the VIRGILE study. The proportion of patients reporting AEs was 60%, which is lower than that reported in the phase III clinical trials (94% in FREEDOMS [4] and 86% in TRANSFORMS [5]), which is perhaps attributable to the observational nature of the study. It should be noted that 248 of the 1055 patients starting fingolimod in the VIRGILE study switched to another DMT over the 3-year period, which complicates attribution of causality. Reporting rates for listed AEs for fingolimod, such as lymphopenia, macular oedema, herpes zoster or basal cell carcinoma, ranged from four patients (0.4%) for basal cell carcinoma to 273 for lymphopenia 273 (24.8%). These rates are consistent with those from the phase III trials [4, 5], but somewhat higher than those reported in the PANGAEA study [17]. Two cases of PML were identified in patients receiving fingolimod, but both had recently switched from natalizumab and these events were attributed to the latter treatment (indeed one patient already had symptoms subsequently attributed to PML at the time fingolimod was started). Cardiovascular monitoring at initiation of fingolimod therapy complied with the prescribing information in all cases. The proportion of patients in whom cardiac rhythm abnormalities were observed at this time was low, suggesting that the recommended cardiac contraindications for fingolimod were being respected and largely protected exposed patients from any cardiac toxicity of fingolimod.
The VIRGILE study also allowed HRQoL outcomes to be documented in a large cohort of patients starting treatment with fingolimod, an aspect that has not been widely studied in previous large drug utilisation studies. We did not observe any significant on-treatment change in MusiQoL scores in either the fingolimod or natalizumab cohorts, using either the MusiQoL or EQ-5D instruments. This suggests that these patients, who were not well controlled before starting fingolimod, consider their health to have been stable over the 3-year period of treatment with fingolimod, which is consistent with the absence of a clinically relevant increase in disability score.
The mean MusiQoL study at inclusion was 67.8 in the fingolimod group, which is very close to the value of 65.8 reported in a previous international study of 1992 adult patients with MS [36]. The data are also consistent with the earlier PERFORMS observational study of fingolimod, performed in the Middle East, which also reported no significant change over a 12-month treatment period [14]. In contrast, in the interventional setting, the EPOC study conducted in North America [35] reported an improvement in HRQoL in patients switching from an injectable first-line DMT to fingolimod, using a generic quality of life measure (SF-36) [37]. The impact of fingolimod treatment on HRQoL in the real-world setting over the long-term is still poorly characterised and further studies in this field are needed to clarify the issue [9]. With respect to natalizumab, a large and significant increase of 11 points in MusiQoL score after 3 years’ treatment has been described in a small French study of 48 patients [38], but we were not able to document such a change in the present much larger study.
The EQ-5D score reported here (0.66) is somewhat higher than would be expected from a previous report of data from France collected in 2007 (EQ-5D score 0.67 for EDSS 3 or less, 0.52 for EDSS 4–5, 0.33 for EDSS 6 or higher) [39], which have been used widely in cost-effectiveness analyses in France (e.g. for dimethylfumarate [40]). This difference may reflect a change in perceptions of burden of illness with the introduction of oral treatments as an alternative to the injectables, which were the only DMTs available in 2007 when the earlier ED-5Q data were collected. It would be interesting to perform a dedicated study to collect utility data from patients prescribed oral DMTs or other recent innovative MS treatments for informing future cost–utility analyses.
The study also included, at the request of the French health authorities, a parallel, independent cohort of 330 patients initiating natalizumab patients to serve as a benchmark. Compared to the previous much larger drug utilisation study of natalizumab in France, performed between 2007 and 2012 (TYSEDMUS) [19], patients initiating natalizumab in the present study were of a similar age and had a similar level of disease activity prior to starting treatment, although the level of disability was somewhat lower (mean EDSS score 3.2 compared to 3.5 in TYSEDMUS). It is not appropriate to compare outcomes between the natalizumab and fingolimod cohorts because they differed in many characteristics that influence outcome, namely prior relapse activity, disability at inclusion and prior DMT history, and indeed this was not the objective of the VIRGILE study. Indeed, the guidelines of the French health authorities for drug utilisation studies specify that comparisons between treatment strategies in such studies should not be made unless the study has been designed specifically to do this with appropriate statistical measures to avoid inclusion bias [41]. A comparative observational study to compare two different DMTs would require a more rigorous methodology, for example by inclusion of cases using propensity score matching to minimise between-group differences at baseline, an approach that has been previously used successfully to compare treatment outcomes between fingolimod and natalizumab [16].
It is also of interest to compare changes in the patterns of care in everyday clinical practice observed in the VIRGILE study (conducted between 2014 and 2019) to those in the earlier GA Observatory (2005–2013) [8] and in the TYSEDMUS study [18, 19]. Compared to these earlier studies, the proportion of patients in VIRGILE who received no further DMT following discontinuation of the study treatment was lower (24%, compared to around 50% in the GA and natalizumab cohorts). This may reflect the wider range of treatment options now available. Persistence rates at 3 years were around 50% both for fingolimod in VIRGILE and for GA [8] (natalizumab discontinuation followed specific stopping rules and the TYSEDMUS data are not thus comparable). In VIRGILE, more than 90% of patients starting fingolimod underwent an MRI evaluation in the following 3 years. This suggests that MRI has now become a standard part of management of patients with MS in France, in contrast to the situation when the GA Observatory was initiated when it was considered that MRI was too rarely performed in everyday practice to be worth collecting data in the study [8].
Conclusions
The VIRGILE study has shown that the effectiveness and tolerability of fingolimod when used in everyday clinical practice over a period of 3 years are consistent with what would be expected from the interventional phase III studies. Fingolimod offers sustained reductions in clinical and radiological disease activity, preserved quality of life and is generally well tolerated. This study highlights the utility of fingolimod as a DMT for the long-term management of patients with MS.
References
Mehling M, Kappos L, Derfuss T. Fingolimod for multiple sclerosis: mechanism of action, clinical outcomes, and future directions. Curr Neurol Neurosci Rep. 2011;11(5):492–7.
Thomas K, Proschmann U, Ziemssen T. Fingolimod hydrochloride for the treatment of relapsing remitting multiple sclerosis. Expert Opin Pharmacother. 2017;18(15):1649–60.
Bordet R, Camu W, De Seze J, Laplaud DA, Ouallet JC, Thouvenot E. Mechanism of action of s1p receptor modulators in multiple sclerosis: the double requirement. Rev Neurol (Paris). 2020;176:100–12.
Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401.
Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15.
Miller AE. Switching or discontinuing disease-modifying therapies for multiple sclerosis. Continuum (Minneap Minn). 2016;22(3):851–63.
Hansen K, Schussel K, Kieble M, et al. Adherence to disease modifying drugs among patients with multiple sclerosis in Germany: a retrospective cohort study. PLoS ONE. 2015;10(7): e0133279.
Lebrun-Frenay C, Moulignier A, Pierrot-Deseilligny C, et al. Five-year outcome in the copaxone observatory: a nationwide cohort of patients with multiple sclerosis starting treatment with glatiramer acetate in France. J Neurol. 2019;266(4):888–901.
Druart C, El Sankari S, van Pesch V. Long-term safety and real-world effectiveness of fingolimod in relapsing multiple sclerosis. Patient Relat Outcome Meas. 2018;9:1–10.
Frisell T, Forsberg L, Nordin N, et al. Comparative analysis of first-year fingolimod and natalizumab drug discontinuation among Swedish patients with multiple sclerosis. Mult Scler. 2016;22(1):85–93.
Lapierre Y, O’Connor P, Devonshire V, et al. Canadian experience with fingolimod: adherence to treatment and monitoring. Can J Neurol Sci. 2016;43(2):278–83.
Bergvall N, Lahoz R, Reynolds T, Korn JR. Healthcare resource use and relapses with fingolimod versus natalizumab for treating multiple sclerosis: a retrospective US claims database analysis. Curr Med Res Opin. 2014;30(8):1461–71.
Bergvall N, Petrilla AA, Karkare SU, et al. Persistence with and adherence to fingolimod compared with other disease-modifying therapies for the treatment of multiple sclerosis: a retrospective US claims database analysis. J Med Econ. 2014;17(10):696–707.
Achiron A, Aref H, Inshasi J, et al. Effectiveness, safety and health-related quality of life of multiple sclerosis patients treated with fingolimod: results from a 12-month, real-world, observational PERFORMS study in the Middle East. BMC Neurol. 2017;17(1):150.
Izquierdo G, Damas F, Paramo MD, Ruiz-Pena JL, Navarro G. The real-world effectiveness and safety of fingolimod in relapsing-remitting multiple sclerosis patients: an observational study. PLoS ONE. 2017;12(4): e0176174.
Guger M, Enzinger C, Leutmezer F, et al. Real-life clinical use of natalizumab and fingolimod in Austria. Acta Neurol Scand. 2018;137(2):181–7.
Ziemssen T, Lang M, Tackenberg B, et al. Real-world persistence and benefit-risk profile of fingolimod over 36 months in Germany. Neurol Neuroimmunol Neuroinflamm. 2019;6(3): e548.
Papeix C, Vukusic S, Casey R, et al. Risk of relapse after natalizumab withdrawal: results from the French TYSEDMUS cohort. Neurol Neuroimmunol Neuroinflamm. 2016;3(6): e297.
Passante N. for the TYSEDMUS STudy Group: TYSEDMUS: suivi observationnel prospectif des patients atteints de sclérose en plaques et traités par TYSABRI® (natalizumab) dans les bases des données EDMUS en France: données finales à cinq ans. Rev Neurol (Paris). 2013;169(Suppl 2):A227.
Haute Autorité de Santé: Guide Méthologique. Études en vie réelle pour l’évaluation des médicaments et dispositifs médicaux. 2011. Available from: Haute Autorité de santé – Service communication et information, 5 avenue du Stade de France, 93218 Saint-Denis, France.
European Medicines Agency. Gilenya 0.25 mg hard capsules; Gilenya 0.5mg hard capsules. Summary of product characteristics. 2011. https://www.ema.europa.eu/en/documents/product-information/gilenya-epar-product-information_en.pdf.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302.
Confavreux C, Compston DA, Hommes OR, McDonald WI, Thompson AJ. EDMUS, a European database for multiple sclerosis. J Neurol Neurosurg Psychiatry. 1992;55(8):671–6.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–52.
Simeoni M, Auquier P, Fernandez O, et al. Validation of the Multiple Sclerosis International Quality of Life questionnaire. Mult Scler. 2008;14(2):219–30.
Rabin R, de Charro F. EQ-5D: a measure of health status from the EuroQol Group. Ann Med. 2001;33(5):337–43.
Moreau T, Bungener C, Heinzlef O, et al. Anxiety and coping strategy changes in multiple sclerosis patients initiating Fingolimod: The GRACE Prospective Study. Eur Neurol. 2017;77(1–2):47–55.
Defer G, de Seze J, Bouee S, et al. Outcomes and treatment management of a French cohort suffering from multiple sclerosis: a retrospective epidemiological study. Mult Scler Relat Disord. 2018;25:276–81.
Vollmer B, Ontaneda D, Harris H, et al. Comparative discontinuation, effectiveness, and switching practices of dimethyl fumarate and fingolimod at 36-month follow-up. J Neurol Sci. 2019;407: 116498.
Ferrè L, Mogavero A, Clarelli F, et al. Early evidence of disease activity during fingolimod predicts medium-term inefficacy in relapsing-remitting multiple sclerosis. Mult Scler. 2021;27(9):1374–83.
Quirant-Sánchez B, Hervás-García JV, Teniente-Serra A, et al. Predicting therapeutic response to fingolimod treatment in multiple sclerosis patients. CNS Neurosci Ther. 2018;24(12):1175–84.
Teniente-Serra A, Hervás JV, Quirant-Sánchez B, et al. Baseline differences in minor lymphocyte subpopulations may predict response to fingolimod in relapsing-remitting multiple sclerosis patients. CNS Neurosci Ther. 2016;22(7):584–92.
Pozzilli C, Prosperini L. Clinical markers of therapeutic response to disease modifying drugs. Neurol Sci. 2008;29(Suppl 2):S211-213.
Rio J, Nos C, Tintoré M, et al. Defining the response to interferon-beta in relapsing-remitting multiple sclerosis patients. Ann Neurol. 2006;59(2):344–52.
Rio J, Rovira A, Tintore M, et al. Evaluating the response to glatiramer acetate in relapsing-remitting multiple sclerosis (RRMS) patients. Mult Scler. 2014;20(12):1602–8.
Fernández O, Baumstarck-Barrau K, Simeoni MC, Auquier P. Patient characteristics and determinants of quality of life in an international population with multiple sclerosis: assessment using the MusiQoL and SF-36 questionnaires. Mult Scler. 2011;17(10):1238–49.
Fox E, Edwards K, Burch G, et al. Outcomes of switching directly to oral fingolimod from injectable therapies: results of the randomized, open-label, multicenter, Evaluate Patient OutComes (EPOC) study in relapsing multiple sclerosis. Mult Scler Relat Disord. 2014;3(5):607–19.
Planche V, Moisset X, Morello R, et al. Improvement of quality of life and its relationship with neuropsychiatric outcomes in patients with multiple sclerosis starting treatment with natalizumab: a 3-year follow-up multicentric study. J Neurol Sci. 2017;382:148–54.
Kobelt G, Texier-Richard B, Lindgren P. The long-term cost of multiple sclerosis in France and potential changes with disease-modifying interventions. Mult Scler. 2009;15(6):741–51.
Chevalier J, Chamoux C, Hammès F, Chicoye A. Cost-effectiveness of treatments for relapsing remitting multiple sclerosis: a French societal perspective. PLoS ONE. 2016;11(3): e0150703.
Haute Autorité de Santé: Guide Méthologique. Études en vie réelle pour l’évaluation des médicaments et dispositifs médicaux. 2021. https://www.has-sante.fr/upload/docs/application/pdf/2021-06/guide_etude_en_vie_reelle_medicaments__dm.pdf. Accessed Feb 2022.
Acknowledgements
The work was performed in collaboration with the Observatoire Français de la Sclérose en Plaques (OFSEP), a national cohort supported by a public grant from the French Agence Nationale de la Recherche within the context of the Investments for the Future programme (reference ANR-10COHO-002; www.ofsep.org). The authors would like to thank all the patients who participated for their valuable contribution to the study.
Virgile Study Group: Abdelhakim ABDELMOUMNI, Corbeil Essonnes; Mouhmmad Jamal AL ALOUCY, Gonesse; Abdullatif AL KHEDR, Amiens; Amer AL NAJJAR CARPENTIER, Orsay; Bernard ALONZO, Marseille; Tony ALTARCHA, Corbeil Essonnes; Amevi ANANIVI, Mont-de-Marsan; Géraldine ANDRODIAS, Lyon; Gilles ANGIBAUD, Montauban; Marie-Sylvie ARTAUD-URIOT, Angoulême; Dominique AUDRY-CHABOUD, Dijon; Marie BARRE, Chambéry; Philippe BARRES, Nice; Rabah BENRABAH, Paris; Eric BERGER, Besançon; François-Xavier BERGOUIGNAN, Bayonne; Patricia BERNADY, Bayonne; Christophe BILLY, Auxerre; Christian BLANCHARD, Brest; Mickaël BONNAN, Pau; Jean-Paul BORSOTTI, Dijon; Catherine BOSSU-VAN NIEUWENHUYSE, Trélazé; Jean-Claude BOUFFETEAU, Saint Quentin; Sophie BOUILLAGUET, Nogent-sur-Marne; Yassine BOUKRICHE, Béziers; Jean-Marc BOULESTEIX, Cahors; Bertrand BOURRE, Rouen; David BRASSAT, Toulouse; Alain BREDIN, Blaye; Bruno BROCHET, Bordeaux; Helene BRUGEILLES BAGUELIN, Le Chesnay; Ousmane CAMARA, Pontivy; Jean-Philippe CAMDESSANCHE, Saint Priest-en-Jarez; William CAMU, Montpellier; Christophe CAREL, Albi; Bertrand CARLANDER, Montpellier; Olivier CASEZ, Grenoble; Giovanni CASTELNOVO, Nîmes; Marie-Pierre CHANEL-SOULIER, Biarritz; Stéphane CHAPUIS, Montluçon; Mirella CIMPOESU, Eaubonne; Jonathan CIRON, Poitiers; Pierre CLAVELOU, Clermont-Ferrand; Christine CLERC, Montbéliard; Renato COLAMARINO, Vichy; Christophe COURATIER, Aix-en-Provence; Sylvie COURTOIS, Mulhouse; Marc COUSTANS, Quimper; Alain CREANGE, Créteil; Antoine DANIELLI, Montauban; Thomas DE BROUCKER, Saint Denis; Jérôme DE SEZE, Strasbourg; Marc DEBOUVERIE, Nancy; Gilles DEFER, Caen; Jérôme DELORME, Lyon; Béatrice DENIS, Marseille; Fayçal DEROUICHE, Mulhouse; Philippe DEVOS, Boulogne-sur-Mer; Anne-Marie DEYROLLE, Saint Palais; Michel DIB, Paris; Joseph DIB, Metz; Eric DIOT, Vienne; Emmanuelle DOURY, Chateaubriant; Sophie DUFOURD-DELALANDE, Tourcoing; Corinne DUPEL-POTTIER, Pontoise; Patrick DUSSAUX, Pontoise; Gilles EDAN, Rennes; Thibault EDOUARD, Saint Aubin-sur-Scie; Jean-Pierre ESCAILLAS, Montluçon; Didier FERRIBY, Tourcoing; Nicolas FOUILLET, Saint Jean-de-Luz; Guillaume FROMAGER, Caen; Tsouria GAIDA-ROSTANE, La Teste-de-Buch; Philippe GAIDA, La Teste; Guillaume GAL, Roanne; Guillaume GARRIGUES, Montpellier; Annick GAYOU-JOYEUX, Dax; Arnaud GENTIL, Dijon; Philippe GERARD, Abbeville; Julien GERE, Chambery; Laurence GIGNOUX, Lyon; Philippe GIRARD, Sète; Pierric GIRAUD, Metz-Tessy; Michel GOUTTARD, Bourg-en-Bresse; Pierre GRAS, Dijon; Anne Marie GUENNOC, Tours; Michel GUGENHEIM, Rambouillet; Laurent GUILLOTON, Mornant; Karim HADJOUT, Rodez; Patrick HAUTECOEUR, Lille; Yawo HEGBE, Mont de Marsan; Olivier HEINZLEF, Poissy; Patrice HENRY, Toulouse; Yann HERVE, Carcassonne; Jihad HIJAZI, Beauvais; Pascale HOMEYER, Aubenas; Bernard HUTTIN, Epinal; Olivier ILLE, Mantes-la-Jolie; Alain JAGER, Thionville; Laurentiu JOMIR, Lyon; Nabil KARDOUS, Sarrebourg; Agnès KEROUANTON, Caen; Comlan Paul KPADE, Le Havre; Christophe KUBLER, Altkirch; Pierre LABAUGE, Montpellier; François LALLEMENT, Saint Brieuc; Nicolas LANDRAGIN, Montpellier; David Axel LAPLAUD, Saint Herblain; Henda LARIBI, Athis-Mons; Gilles LAVERNHE, Gap; Pierre-Éric LE BIEZ, Cherbourg-Octeville; Françoise LE BRAS, Niort; Patrick LE COZ, Arras; Christine LEBRUN-FRENAY, Nice; Josette LECHE, Vendôme; Sara Julia LEDER, Paris; Alain LEGOUT, Le Mans; Michele LEVASSEUR, Orsay; Alberta LORENZI-PERNOT, Mornant; Pierre LOUCHART, Douai; Fabien LOUILLET, Saint Aubin-lès-Elbeuf; Laurent MAGY, Limoges; Sophie MAILLARD, Saint Dizier; Elisabeth MAILLART, Paris; Marcel MAILLET-VIOUD, Montluçon; Catherine MALLECOURT-EMBERGER, Toulon; Éric MANCHON, Gonesse; Alexandre MANIA, Béziers; Laurent MARTINEZ-ALMOYNA, Aix-en-Provence; Mikel MARTINEZ, Dax; Serge MASSENGO, Lannion; Dominique MAUGIN, Trélazé; Souraya MEDJBEUR, Villejuif; Gayané MELIKSETYAN, Argenteuil; Michael MENASSA, Dijon; Dalia MESHAKA-DIMITRI-BOULOS, Créteil; Gérard MICK, Voiron; Thibault MOREAU, Dijon; Antoine MOULIGNIER, Paris; Isabelle MOURAND, Montpellier; Jean-Philippe MULLER, Antibes; Philippe NEUSCHWANDER, Lyon; Argentino NIBBIO, Boulogne-sur-Mer; Chantal NIFLE, Le Chesnay; Jean-Bertin NKENDJUO, Dunkerque; Ghislain NOKAM TALOM, Deauville; Sophie ORY, Lorient; Ivania PATRY, Corbeil-Essonnes; Bernard PEDESPAN, Agen; Jean PELLETIER, Marseille; Delia-Gianina PENCU, Romans-sur-Isère; Bruno PERROUTY, Carpentras; Stéphane PEYSSON, Gleizé; Irène POPA-COMAN, Bobigny; André POULIQUEN, Saint-Aubin-sur-Scie; Christophe PRAT, Saint Michel; Adriana PRUNDEAN, Angers; Fataï RADJI, Agen; Haja Tiana RAKOTOHARINANDRASANA, Dreux; Lilia RAZLOG, Soissons; Philippe REMY, Créteil; Christophe ROBIN, Roanne; Gilles RODIER, Metz-Tessy; Jérôme ROMERO, Fréjus; Brigitte ROUALDES, Créteil; François ROUHART, Brest; Irene RUGGIERI, Marseille; Feras Abdul SAMAD, Châtellerault; Irina SARAFIANT, Saint Avold; Stephane SCHAEFFER, Caen; Nicolas SCHMIDT, Rueil-Malmaison; Philippe SCHUERMANS, La Rochelle; Nicolas SEILLER, Sarreguemines; Thierry SOISSON, Orleans; Annie SORTAIS, Laval; Bruno STANKOFF, Paris; Sabrina STEFANIZZI-DEBUC, Meaux; Laurent SUCHET, Marseille; Jean TARDY, Cornebarrieu; Gregory TAURIN, Saint Malo; Florent THABUY, Créteil; Marie THEAUDIN, Le Kremlin Bicêtre; Caroline TILIKETE-FROMENT, Bron; Ayman TOURBAH, Reims; Patricia TOURNIAIRE, Avignon; Sylvie TREFOURET, La Seyne-sur-Mer; Michel VASTENE, Castelnau-le-Lez; Pierre VERDURE, Saint Aubin-lès-Elbeuf; Patrick VERMERSCH, Lille; Frédérique VIALA, Toulouse; Dorothée VIDET-GIBOU, Vannes; Elisabeth VIDRY, Albi; Jean-Marc VISY, Reims; Sandra VUKUSIC, Bron; Marc WAGNER, Ars Laquenexy; Valery WATTIER, Saint Julien-en-Genevois; Christophe ZAENKER, Colmar; François ZIEGLER, Vesoul; Jean-Médard ZOLA, Verdun.
Funding
Novartis Pharma S.A.S., Rueil-Malmaison, France funded the conduct of the study, medical writing support and the Rapid Service Fee for publication.
Medical Writing Assistance
Medical writing services for this article were provided by Dr. Adam Doble, SARL Foxymed, Paris, France, funded by Novartis Pharma S.A.S., Rueil-Malmaison, France.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article and take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Bashar Allaf, Isabelle Chouette, Marc Coustans, Marc Debouverie, Christine Lebrun-Frenay, François Heintzmann, Gisela Kobelt, Emmanuelle Leray, Fabienne Lamy, Caroline Papeix and Jean-Marc Visy contributed to the design of the study and the preparation of the protocol. Bashar Allaf, Barbara Durand and Fabienne Lamy supervised the conduct of the study. Driss Kamar supervised the data analysis. Giovanni Castelnovo, Isabelle Chouette, Barbara Durand, Marc Coustans, Marc Debouverie, Christine Lebrun-Frenay, Emmanuelle Grevat, François Heintzmann, Driss Kamar, Fabienne Lamy, Pierre Levy, Caroline Papeix, Eric Raponi and Jean-Marc Visy contributed to the interpretation of the data and the preparation of this manuscript.
Prior Publication
Some of the data provided in the present article was presented as a poster at the 37th Congress of the European Committee for Treatment and Research in Multiple Sclerosis, 13 September 2019, Stockholm, Sweden: Abstract No. P1388 (https://onlinelibrary.ectrims-congress.eu/ectrims/2019/stockholm/278589/giovanni.castelnovo.long-term.effectiveness.safety.and.tolerability.with.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D2%2Asearch%3Dlebrun-frenay).
Disclosures
Giovanni Castelnovo has received consultancy fees from Merck Serono, Biogen Idec, Novartis, Teva, Bayer, Merz, Ipsen, Genzyme, Allergan. Marc Coustans reports personal fees from Biogen and personal fees from Teva Pharma, Novartis and Genzyme. Emmanuelle Leray reports research grants from the French National Agency for Medicines and Health Product Safety (ANSM), the EDMUS Foundation and the ARSEP Foundation, as well as consulting and lecture fees or travel grants from Biogen, Genzyme, MedDay Pharmaceuticals, Merck Serono, Novartis and Roche. Caroline Papeix has received fees as a consultant, advisory board member or speaker for Roche, Novartis, Biogen, Teva, Sanofi, Genzyme. Christine Lebrun-Frenay has received consultancy fees from Novartis, Roche and Genzyme. Gisela Kobelt has received fees for projects and consultancy from Almirall, Biogen, Bayer Schering, BMS, Genzyme, Medday, Merck, Novartis, Pfizer, Roche, Sanofi, Teva. Pierre Levy has received consultancy fees from AbbVie, Bayer, Actelion, Amgen, Astellas, Bayer, Biogen, BMS, Boehringer Ingelheim, Daïchi-Sankyo, Gilead, GSK, Hospira, Janssen, MSD, Novartis, Novo Nordisk, Pfizer, Roche, Sanofi. Jean-Marc Visy has received fees from Novartis for educational activity and consulting. Marc Debouverie has received fees for consulting, research and workshops for Biogen, Bayer Schering, Genzyme, Merck, Novartis, Sanofi and Teva. Fabienne Lamy, François Heintzmann, Bashar Allaf, Barbara Durand, Emmanuelle Grevat, Eric Raponi and Isabelle Chouette are employees of Novartis Pharma, France. Driss Kamar is an employee of Ividata Life Science, mandated by Novartis Pharma, France for statistical advice on the study.
Compliance with Ethics Guidelines
The study was conducted in accordance with the Declaration of Helsinki and with all other relevant international and French legislation and followed Good Clinical Practice. The protocol was submitted for approval to the CPP (Comité de Protection des Personnes; ethics committee), and the opinion of the CPP was transmitted to the French Safety of Medicines Agency and to the CCTIRS (Comité Consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé), as required under French law for observational studies. Approval (No. 13.307 dated 22 May 2013) was at a national level and covered all participating centres. Prior to inclusion, each eligible patient was provided with a study information leaflet and was given the opportunity not to participate in the study. Patient consent to the collection, analysis and potential monitoring of the data required for the study was given and documented in the study database. Patient enrolled through OFSEP also signed the OFSEP consent form. All patient data was anonymised before entry into the database. The study protocol was submitted to the French Data Protection Agency (Commission Nationale de l’Informatique et des Libertés; CNIL) for comment before initiation of the study.
Data Availability
Anonymised data will be made available to qualified external researchers, with requests reviewed and approved by an independent review panel on the basis of scientific merit.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The members of The Virgile Study Group are listed in the Acknowledgements section.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Papeix, C., Castelnovo, G., Leray, E. et al. Long-Term Effectiveness, Safety and Tolerability of Fingolimod in Patients with Multiple Sclerosis in Real-World Treatment Settings in France: The VIRGILE Study. Neurol Ther 11, 633–658 (2022). https://doi.org/10.1007/s40120-022-00334-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-022-00334-y